
本文作者:杉杉
导读
近日,南京大学朱少林教授课题组在Angew. Chem. Int. Ed.上发表论文,通过使用NiH催化剂和新型手性双咪唑啉配体,成功实现了烯基芳烃与芳基碘的高对映和区域选择性还原氢芳基化反应。同时,该反应可在极其温和的条件下,以优异的收率和对映选择性获得多种对映体富集的1,1-二芳基烷烃衍生物,此结构存在于多种生物活性分子中。
Enantio- and Regioselective NiH-Catalyzed Reductive Hydroarylation of Vinylarenes with Aryl Iodides
Yuli He, Chuang Liu, Lei Yu, and Shaolin Zhu
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202010386
正文
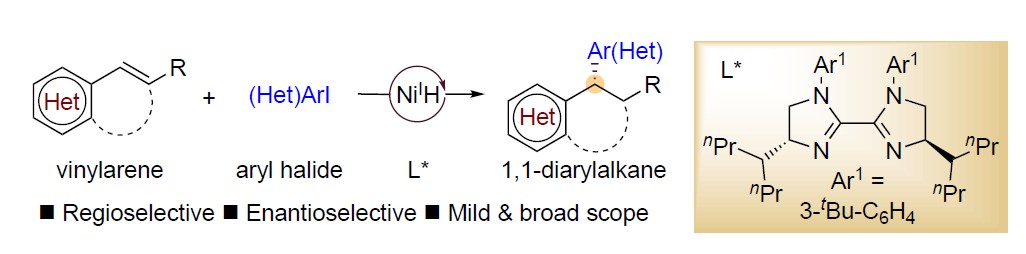
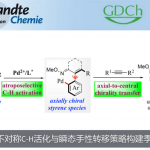
在过去的二十年中,由于镍催化具有原子经济性、易获得多种氧化态等优点,从而使其广泛应用于交叉偶联和还原偶联反应中。早期,主要集中在两个经典交叉偶联底物的ipso-偶联上。最近,使用易获得的烯烃作为起始原料,通过NiH催化的还原性加氢官能化或还原性迁移加氢官能化,可将各种官能团引入其近端或远端的位置。这种通用的合成方法避免了早期方法中有机金属试剂的产生,并具有控制区域和立体选择性的潜能。同时在该过程中,可在新成键的两个碳原子上建立sp3-杂化的立体中心(Figure 1a)。最近,加州理工学院的Fu课题组和中国科学技术大学的Fu课题组以及朱少林教授课题组证明,可以通过对映体收敛的方式(enantioconvergent fashion),来控制外消旋烷基亲电试剂碳立体中心的产生(i in Figure 1a)。然而,对于非手性烯烃的碳立体中心的控制仍然具有挑战性(ii in Figure 1a)。

过渡金属催化不对称氢芳基化反应,是构建芳基取代手性中心的有效方法,已引起人们的极大兴趣。以芳基亲核试剂和苯乙烯为底物,以醇作为氢来源,Sigman开发了不对称Pd(II)H催化的氢芳基化体系。而Zhou和Mei课题组报道了一个强大的Ni(II)H体系,可实现相同的目标(i in Figure 1b)。然而,在这些反应中,烯烃的范围限于β-未取代的底物。为了克服这些限制,Buchwald课题组报道了Cu(I)H/Pd(II)-协同催化的不对称氢芳基化体系,该体系实现了各种苯乙烯和易于获得的芳基卤化物的反应。前期,朱少林教授课题组使用还原性Ni(I)H体系,实现了具有苄基区域选择性的烯烃的迁移氢芳化反应,同时,作者设想是否立体化学选择性也可同时得到控制,获得对映体富集的1,1-二芳基烷烃(ii in Figure 1b)。在此,南京大学朱少林教授课题组报道了一种温和而稳健的策略,使用易得的双咪唑啉-镍配合物作为唯一催化剂,实现了此类反应。
对于不对称氢芳基化,作者提出了可能的反应过程(Figure 2)。首先,L*NiIH配合物(I)插入到烯烃(1)中,生成烷基镍中间体(II),与芳基碘(2)经氧化加成得到高价的Ar-Ni(III)-烷基中间体(III)。随后,Ar-Ni(III)-烷基配合物(III)可以通过Ni-C键的均裂快速平衡形成Ni(II)/苄基自由基对,若苄基(V)与Ar-Ni(II)-I配合物(IV)的重组具有高对映选择性,则该立体诱导过程将形成对映体富集的Ar-Ni(III)-烷基配合物(III)。最后,配合物(III)经不可逆的还原消除,形成对映体富集的1,1-二芳基烷烃产物(3)和L*NiI-I(VI)。此外,L*NiI-I(VI)可与KF进行配体交换,形成L*NiI-F(VII),然后用化学计量的氢硅烷试剂进行转金属化,以再生L*NiIH配合物(I),从而完成催化循环。

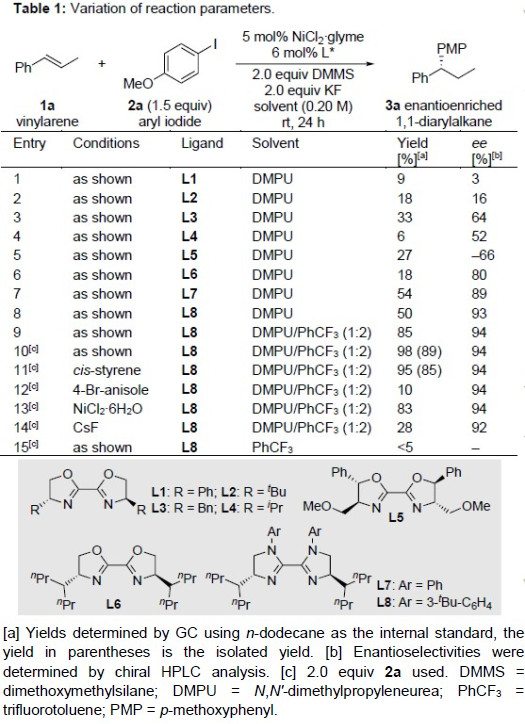
首先,作者以反式-β-甲基苯乙烯1a和4-碘苯甲醚2a作为模型底物,对配体、溶剂等反应条件进行了筛选(Table 1)。使用6 mol%的L8作为配体,5 mol%的NiCl2·glyme作为催化剂,2个当量的二甲氧基(甲基)硅烷(DMMS)作为氢源,2个当量的KF作为碱,同时使用DMPU和PhCF3的混合溶剂,作为最佳反应条件。
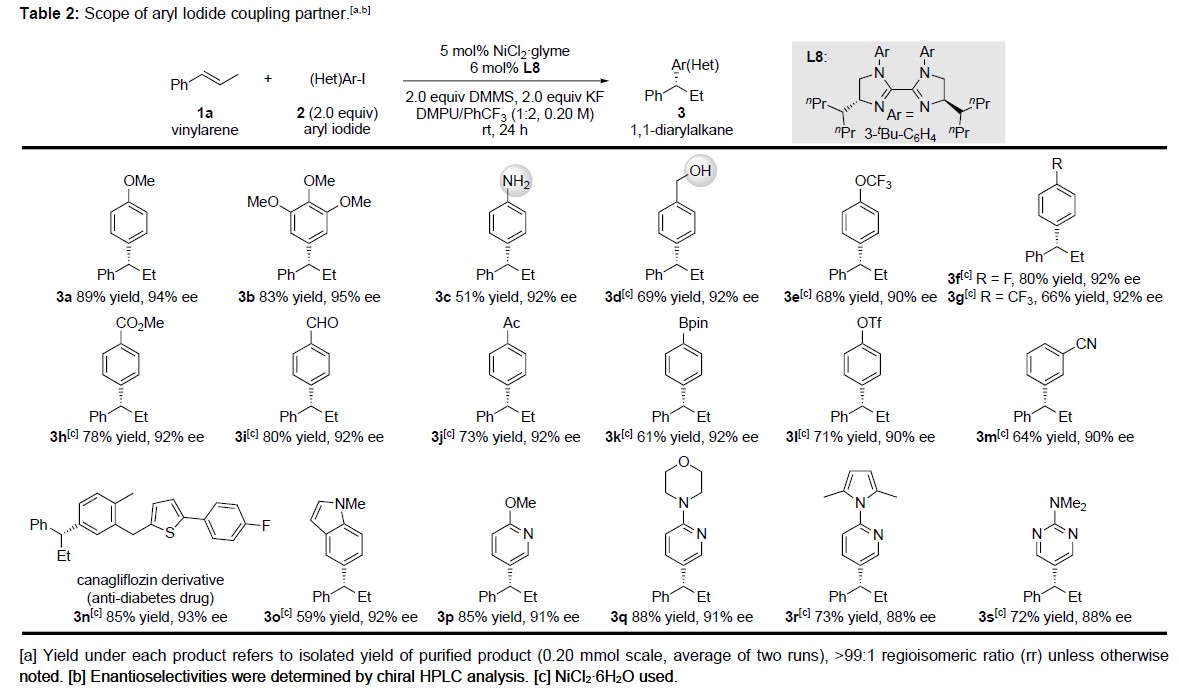
在获得上述最佳反应条件后,作者开始对芳基碘底物2进行了拓展(Table 2)。反应结果表明,富电子的(2a–2d)和吸电子的(2e–2m)芳基碘化物均与体系兼容。同时,各种取代的官能团也能够很好地反应,如醚(2a,2b和2e)、氟化芳基(2f)、三氟甲基(2i)、酯(2h)和腈(2m)。值得注意的是,一些敏感的官能团,如未保护的伯胺和醇(2c–2d)、易还原的醛酮(2i–2j)以及硼酸频哪醇酯(2k)和三氟甲磺酸酯(2l)均能够保持完整。此外,噻吩(2n)、吲哚(2o)、吡啶(2p,2q和2r)和嘧啶(2s)等杂环也同样取得较好的结果。
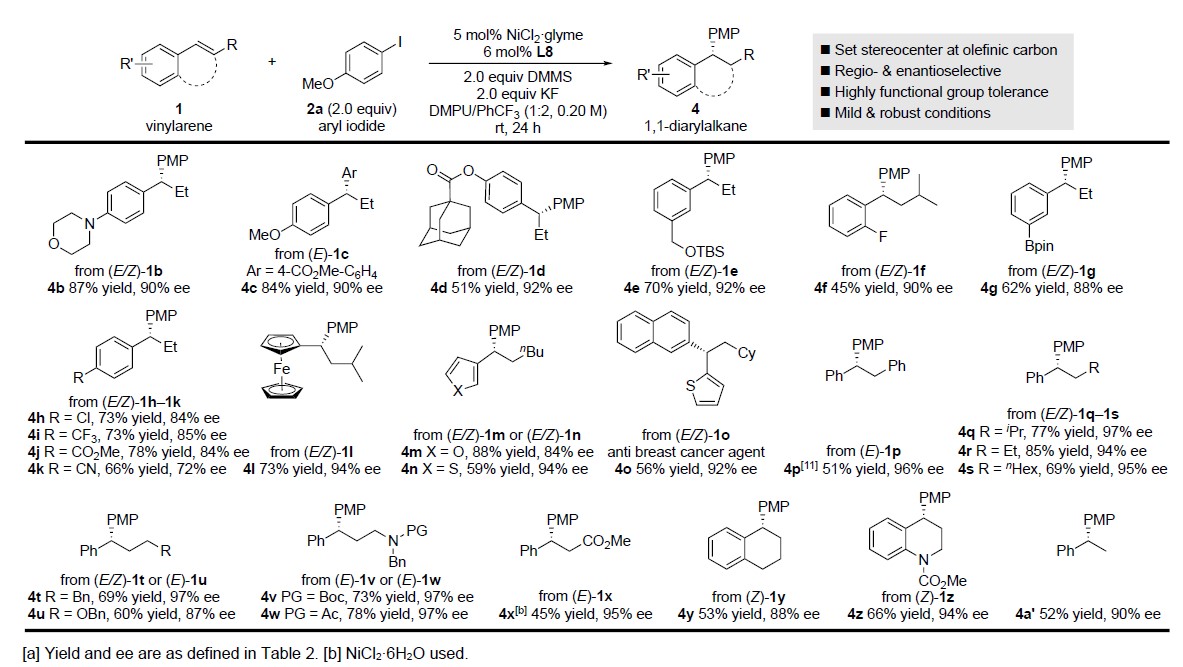
随后,作者对芳基烯烃底物1进行了相关拓展(Table 3)。芳环上具有富电子(1b–1d)和缺电子(1f–1k)基团的底物,均平稳地进行不对称氢芳基化反应。通常,具有富电子(1b–1d)或电子中性(1e)取代基比缺电子(1f–1k)取代基的底物对映选择性高,可能与反应过程中相应苄基的稳定性有关。同时,邻位取代的苯乙烯(1f)、杂芳烃(如呋喃(1m)或噻吩(1n,1o))等,均与体系兼容。而在β-位上带有不同取代基,如芳基(1p)和烷基(1q–1t),或在烷基链的另一端带有杂原子取代基(1u–1w,1z)均能很好地反应。此外,β-未取代苯乙烯的不对称氢芳基化同样有效(1a’)。
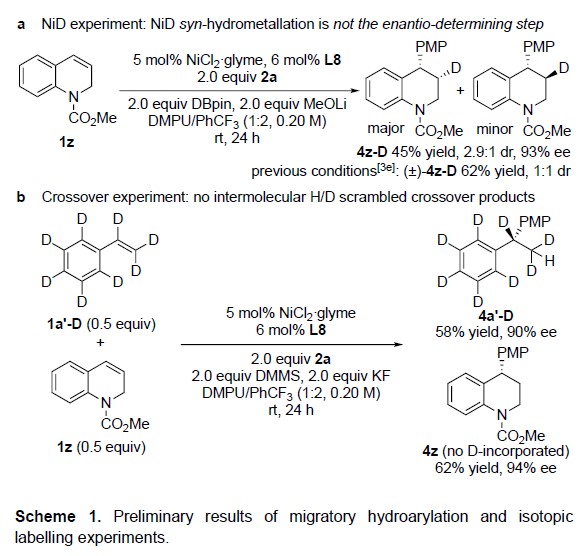
为了进一步了解反应的机理,作者进行了相关的氘代实验(Scheme 1)。首先,在环状苯乙烯(1z)中,加入氘代频哪醇硼烷和4-碘苯甲醚(2a),若NiD与芳基烯烃的顺式氢金属化为对映选择性决定步骤(与CuH反应类似),则会形成非对映体纯的4z–D。但是,观察到在β-位引入氘时,仅以dr = 2.9:1生成4z–D,表明NiD物种仅以中等对映选择性插入到苯乙烯中。当在类似条件下使用非手性配体时,检测到两种4z–D非对映体的量大致相等(dr = 1:1),从而支持了作者提出的机理: Ni(III)到Ni(II)与苄基自由基的快速均裂是形成苄位立体中心的对映选择性决定步骤(Scheme 1a)。此外,为了确定NiH/NiD物种向芳基烯烃的顺式氢金属化是否可逆,作者使用氘标记的苯乙烯(1a’-D)和未氘化的苯乙烯(1z)的1:1混合物进行交叉偶联实验,并未观察到H/D交叉产物,这一结果与最初机理中不可逆的顺式氢金属化步骤一致(Scheme 1b)。
总结
南京大学朱少林教授课题组通过使用NiH催化剂和新型手性双咪唑啉配体,成功实现了烯基芳烃与芳基碘的高对映和区域选择性还原氢芳基化反应,获得多种对映体富集的1,1-二芳基烷烃衍生物。同时,该反应具有底物范围广泛、官能团耐受性良好、反应条件温和等优点。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!











No comments yet.