
本文作者:竹悠
导言
制备出手性分子的两个异构体是有机合成、药物化学及材料科学中的基础任务。因天然来源的手性化合物并不同时具备两个构型,因此要达到这个目的,需要在反应体系中逆转手性结构得到另一构型的产物。本文作者另辟蹊径,通过一种铱Ir催化剂进行两种动力学拆分反应,通过控制反应时间,合成了两个异构体,这种策略称为对映发散性合成。合理安排单个反应,根据反应速率在不同的反应时间淬灭,可以分离出两种截然不同的手性产物,这种方法成为一次性制备两种构型的新策略。
正文
生命体拥有纯手性环境,手性分子的不同构型经常有不同的生理活性,如何制备手性分子的两个构型是有机合成的基础任务。常规的制备光学纯化合物的方法有不对称合成,或者选择一个光学纯构型的手性化合物为起始原料进行合成。这些手性起始化合物有萜类、氨基酸、糖类、生物碱等,它们廉价易得,通常统称为手性池化合物。无论是手性池起始物料还是不对称合成中的辅助物、试剂、催化剂及配体,都要手性结构作为工作的基石,如图1a所示。但这些来源于天然产物的手性化合物往往只有一种构型,因此怎样合成另一个截然不同的构型成为一个难题。
在不对称合成中,反应时间也是个重要参数,特别是在动力学拆分Kinetic resolution (KR)中,利用反应速率的不同,一种构型参与反应,另一种一种构型不反应或反应速率很慢,产物不同从而将两者分离。但即使反应彻底,最高只有50%的转化率。还有种策略为平行动力学拆parallel kinetic resolution(PKR),两个构型分别发生不同的反应,各生成对应的产物,进行分离。虽然动力学拆分和时间有关,但手性试剂或催化剂起决定性作用。
最近上海有机所的游书力课题组,开发了对映发散性合成策略,在同一个催化剂体系下,仅仅依靠反应时间的不同,得到两种构型的产物,如图1b所示,相关论文发表在Nature Chemistry上。
“Hang-Fei Tu, Pusu Yang, Zi-Hua Lin, Chao Zheng, Shu-Li You.
Time-dependent enantiodivergent synthesis via sequential kinetic resolution
Nature Chemistry, ASAP, DOI: 10.1038/s41557-020-0489-1”
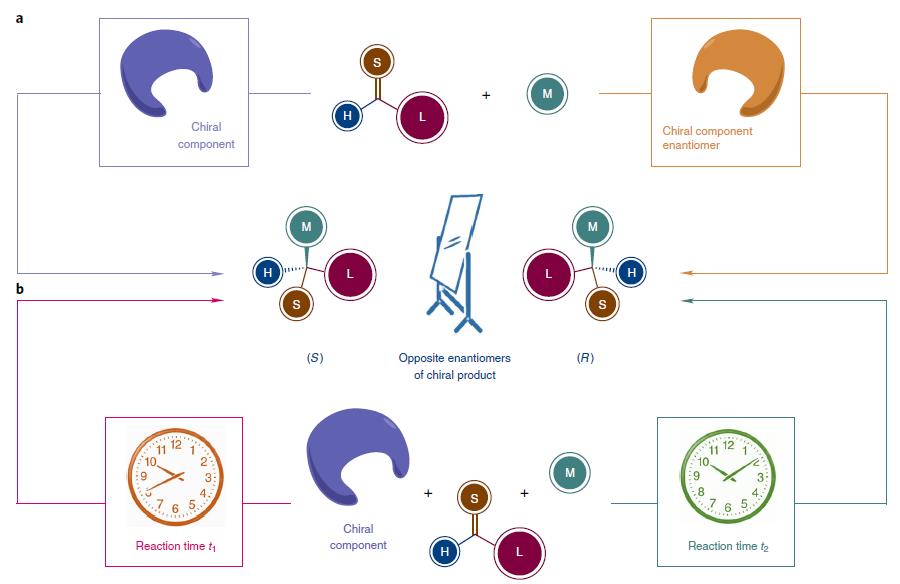
图1. 不同的合成手性异构体策略
结果和讨论
根据早期研究5-或7-羟基喹啉分子内不对称胺基烷基化反应的经验,以6-羟基异喹啉(1a)和叔丁基(1-苯基烷基)碳酸酯(rac-2a)反应作为模板反应,在铱催化剂(Ir(cod)Cl)2、手性配体(S)-L1和添加剂3,5-二氯苯甲酸作用下,以甲醇为溶剂,胺化反应在室温下平稳进行。反应10h后,胺化产物(R)-3aa的收率为78%(98% e.e.),但没有质子酸时,反应仅6min后就发生淬灭反应,得到构型相反的(S)-3aa,收率80%(94% e.e.),产物的构型和选择性与反应时间相关,如图2所示。
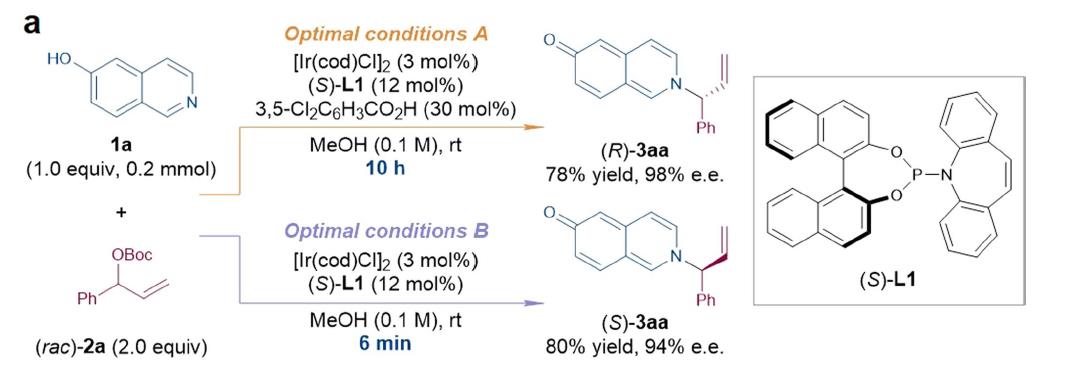
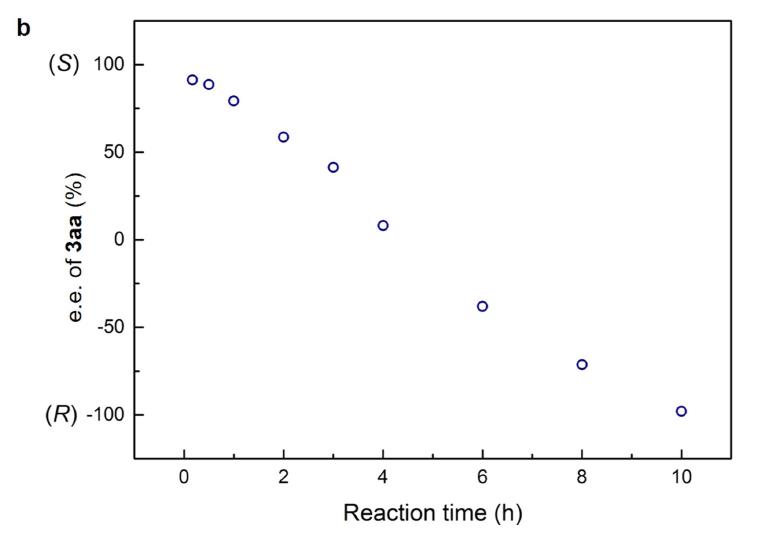
图2a. 时间控制的不同胺化产物构型
这种有趣的实验现象,背后必有隐藏的机理,作者通过一系列对照实验,逐步揭示了真相。
对照实验1:(S)-3aa (93% e.e.) 与2.0 equiv的 (rac)-2a反应,10h后得到(R)-3aa收率75% (88% e.e.)和(S)-4a收率53% (90% e.e.)。如图2b所示,说明产物3aa可以在反应条件A下进行发生取代反应生成(S)-4a,在甲醇中发生了动力学拆分反应。

图2b. (S)-3aa与(rac)-2a在条件A下的反应
对照实验2:消旋体和两个光学纯的2a,分别在条件A下反应。消旋的(rac)-2a在6min淬灭反应时,(S)-3aa的收率为41%(93% e.e.),并回收了43%的(R)-2a(97% e.e),这个动力学拆分反应的选择因子(s)为116,而两个光学纯的2a的收率和选择性都很高,如图2c所示。

图2b. (rac)-2a、(S)-2a和(R)-2a在条件A下的反应
对照实验3:化合物(R)-3aa在(S)-L1的催化下,反应很慢。但换为(R)-L1时,反应非常彻底,收率99%(99% e.e.),动力学拆分反应的选择因子(s)为70。化合物(R)-3aa在(S)-L1的催化下,反应很慢,但换为(R)-L1时,反应却非常彻底,收率99%(99% e.e.),如图3d所示。
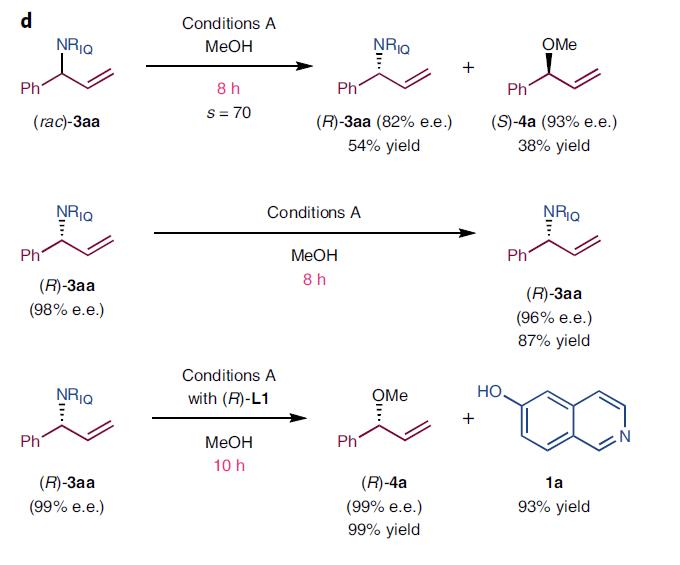
图2c. (rac)-3aa、(R)-3aa在条件A下的反应
用F谱研究氟取代的化合物反应初始速率,(S)-3ab的生成速率最快,对映异构体(R)-3ab与其数量级一样,但慢约3.5倍。(S)-3ab继续反应的速率比(R)-3ab高两个数量级,如图3a所示。通过这个实验可以证明,通过两个连续的动力学拆分,仅控制反应时间作为主要参数,就可以分别制备烷基胺的两个异构体。
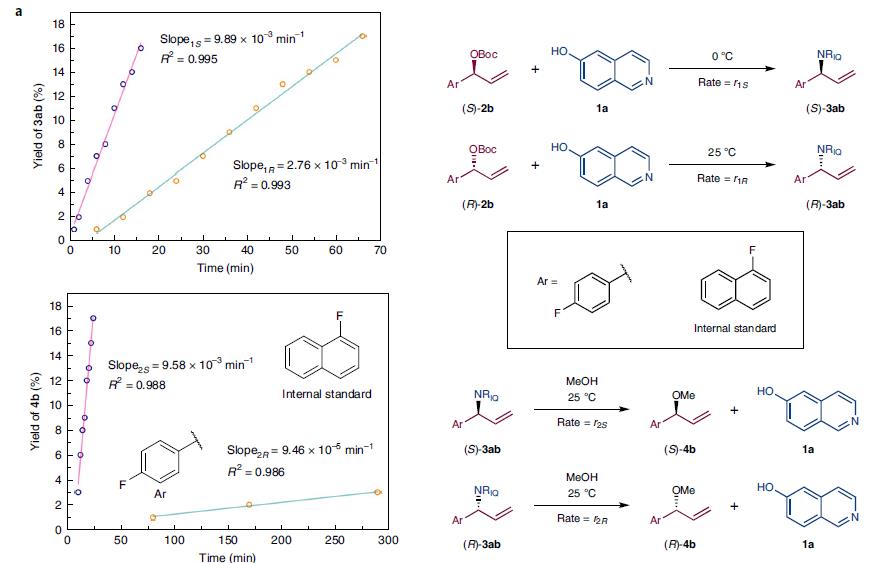
图3a. 用F谱研究氟取代的化合物反应初始速率
图3b更形象的描述这个规程,首先是(rac)-2a在铱催化剂下发生一次动力学拆分,与6-羟基异喹啉发生不对称的烯丙基胺化反应。仅6min后(S)-2a 转化为(S)-3aa,而此时(R)-2a基本未反应,随时间进行(S)-3aa继续反应生成(S)-4a,甲醇作为亲核试剂取代原来的NRIQ。而(R)-3aa逐渐生成累积,它被甲醇取代的速率更慢,因此更加稳定,在10h时为主要产物。
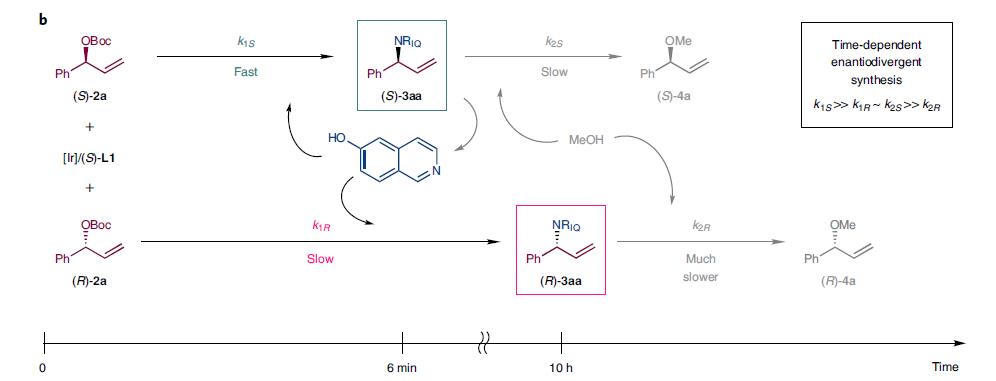
图3b. 反应速率不同,不同时间得到不同产物.
将条件优化后,作者对底物进行了拓展,含卤素、甲基、甲氧基及其它的肉桂基碳酸酯都可以和6-羟基异喹啉反应,收率中等到优秀,立体选择性中R构型的e.e.一般>97%,S构型的e.e.一般>90%。不同结构的异喹啉也有类似的结果,典型结构和收率见下图4
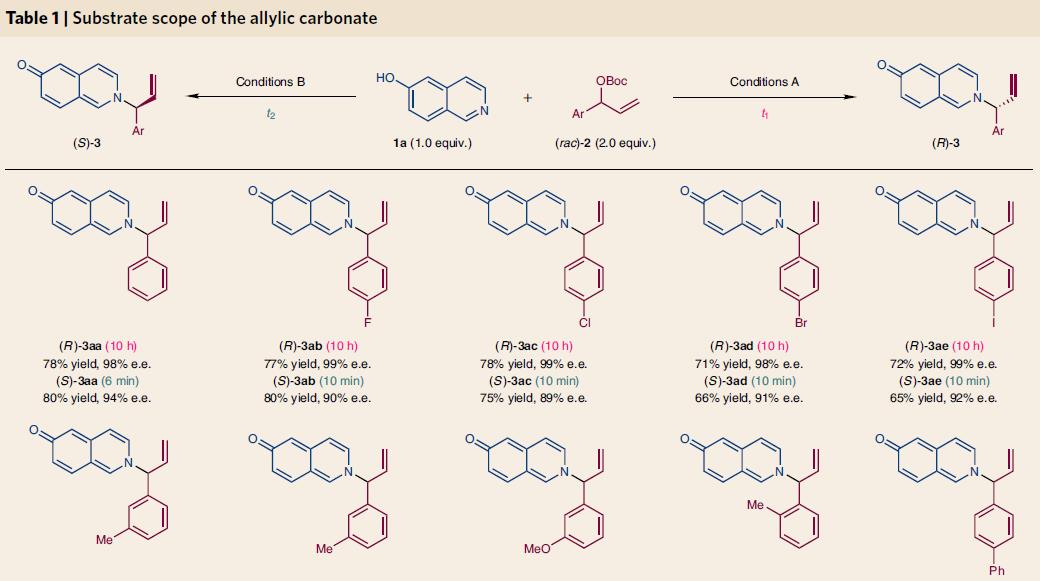
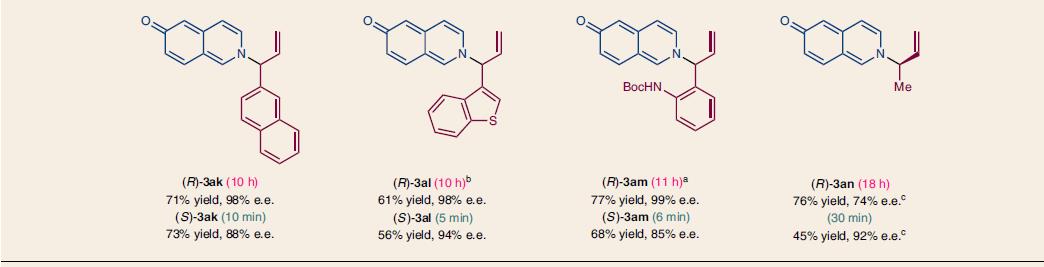
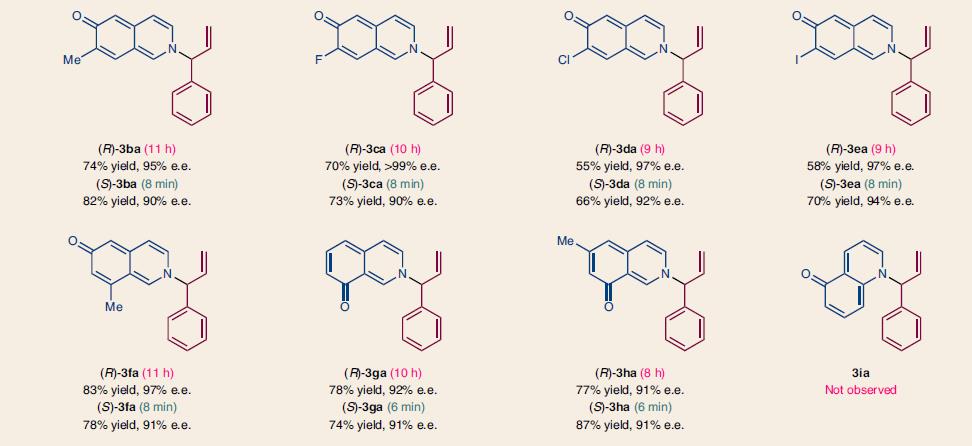
图4. 不同底物情况下的结构和收率
总结
作者通过反应设计,一种催化剂依次促进两种动力学拆分反应,合理安排反应时间,时间一到淬灭反应后,分离出一种对映异构体富集的形式,再延长反应时间,得到另一个异构体。这种新颖的合成策略,仅仅通过反应时间就能控制立体化学,为制备两种不同构型的手性化合物提供了新选择。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.