
本文作者:Sunny华
导读
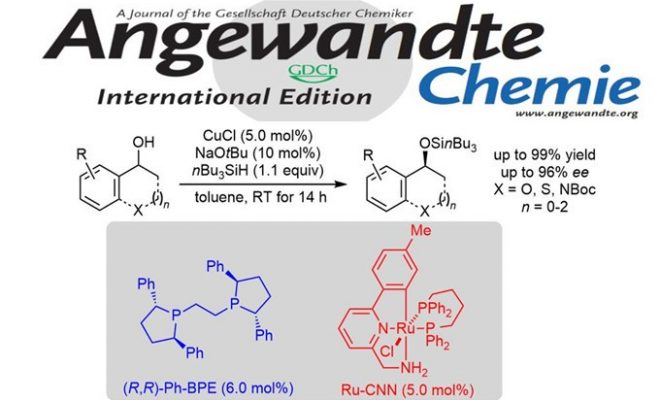
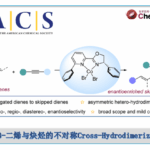
德国柏林工业大学Martin Oestreich教授(主页)等人利用手性铜催化剂和双功能化钌络合物催化剂成功实现了环状及非环状苄醇类化合物的非酶促动态动力学拆分。该反应中,对映选择性Cu-H催化下的Si-O脱氢偶联和过渡金属催化下的底物迅速消旋化过程完美正交兼容,最终以高产率和高对映选择性获得了单一构型产物。日前,相关成果以“热点论文(Hot Paper)”形式在线发表于著名高水平化学期刊——德国应用化学。
“Dynamic Kinetic Resolution of Alcohols by Enantioselective Silylation Enabled by Two Orthogonal Transition-Metal Catalysts”
Jan Seliger and Martin Oestreich*
Angew. Chem. Int. Ed. 2020, ASAP. DOI: 10.1002/anie.202010484 https://doi.org/10.1002/anie.202010484
正文
研究背景
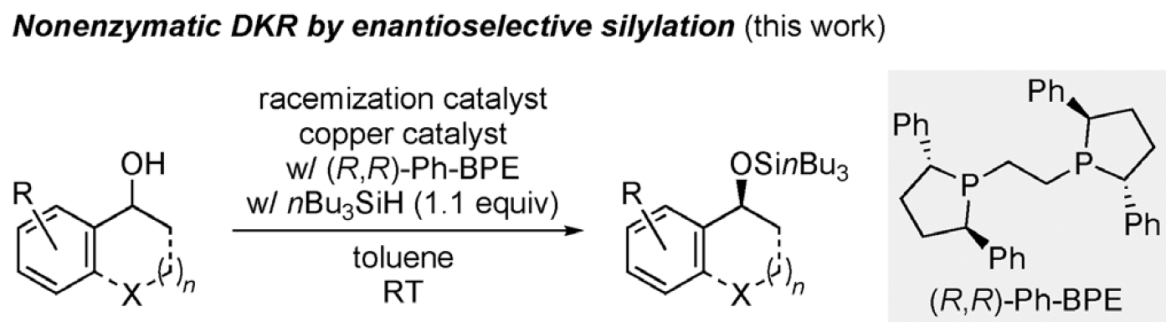
动态动力学拆分(Dynamic Kinetic Resolution,DKR)是获得手性纯醇类化合物的重要方法,它相对于传统动力学拆分(Kinetic Resolution,KR)显著的优点是通过起始原料“原位”迅速的消旋化过程克服了动力学拆分中单个异构体理论最高50%产率的限制。Williams等人率先通过催化转移氢化的方式可以实现醇的催化消旋化[1],随后Bäckvall等人发现钌络合物1是一种高效的醇类化合物消旋化催化剂并且他们对其催化消旋化的机理进行了较为详细的研究,当钌催化剂1与南极假丝酵母脂肪酶B(Candida antarctica lipase B)组合后利用醋酸异丙烯酯作为酰化试剂,他们实现了一系列结构多样化仲醇的化学酶促动态动力学拆分[2]。几年后,Fu等人再次利用钌催化剂1和另一个基于二茂铁骨架的平面手性DMAP衍生物作为亲核活化酰基源的催化剂成功实现了仲醇的非酶促动态动力学拆分[3],这也是迄今为止唯一的非酶促拆分方法。
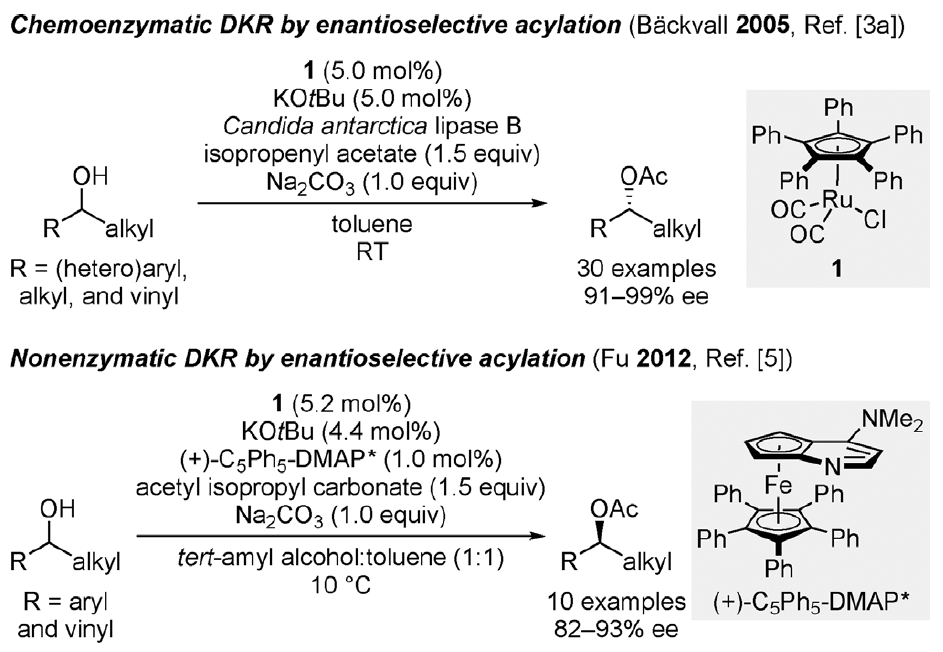
图 1 已报道过的仲醇动态动力学拆分方法(图片来源于Angew)
通过DKR过程引入的酰基通常被脱保护释放出羟基或者作为保护基参与后续反应;硅醚作为羟基保护基具有两个显著优势,除了保护和脱保护过程比较容易外,硅基还具有正交性并且可以调控其稳定性。然而,不同于大量的酶促或非酶促作用下的对映选择性酰基化拆分方法,基于硅基化的拆分方法直到最近20年才得以发展,因此人们对于硅基化方法能否实现DKR更是知之甚少。要想实现这一过程必须满足以下条件:首先拆分体系必须和消旋化催化剂兼容并且消旋化过程速率应远超低活性异构体与拆分试剂作用的速度;其次消旋化催化剂必须不能引起产物的消旋化以及促进非对映选择性的硅基化过程(背景反应)。
研究思路
1.消旋化催化剂的筛选
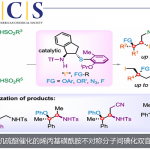
作者首先对多种钌、锇络合物作为潜在的消旋化催化剂进行了研究,正如预期那样,钌络合物1和NaOtBu的组合可以使(S)-2a迅速的消旋化(entry 1)。然而,一旦将已经建立的拆分条件引入,消旋化的效果就急转直下(entries 2-4),这一现象在另一种钌催化剂3a中也观察到了,并且调节催化剂中配体的电子云密度也没有明显改观(entrie 5-7)。于是,作者开始尝试类型不同的双功能化催化剂(4a,4b和5)并且发现它们都具有很高的催化消旋化活性以及对拆分条件具有很好的兼容性(entries 8-10)。
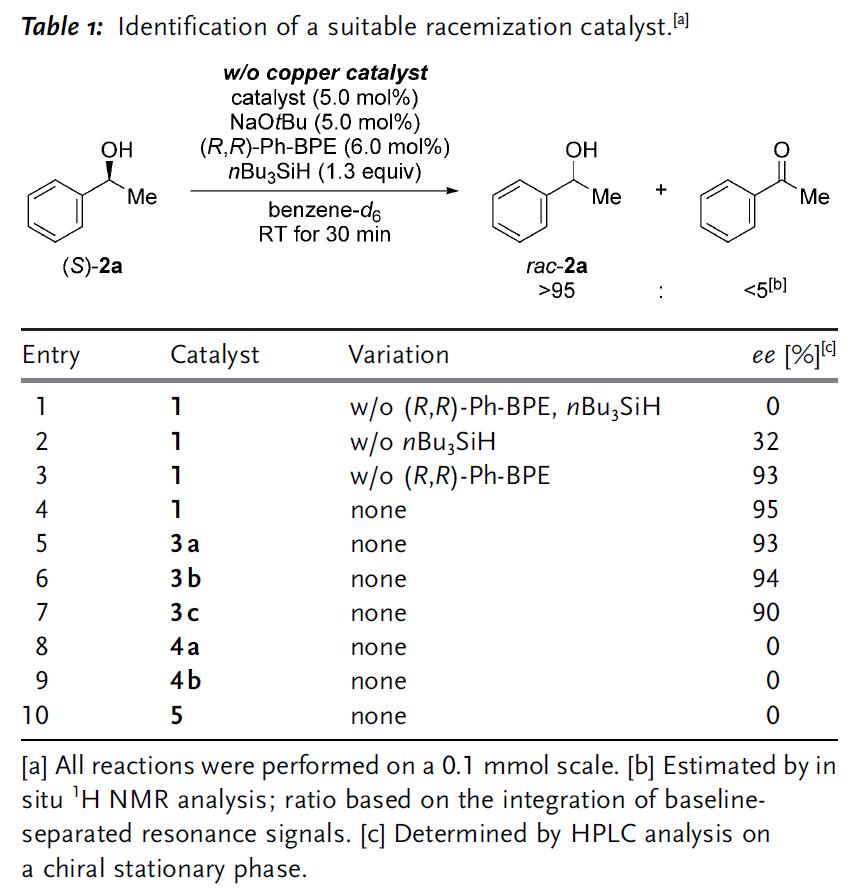
图 2 消旋化催化剂的筛选(图片来源于Angew)
2.背景反应的探究
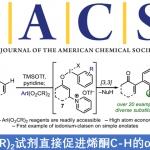
获得上述三个较为理想的消旋化催化剂后,作者随后对三个催化剂的背景反应进行了探究并由此发现三个催化剂在碱性条件下都不会引起产物硅醚(S)-6a的消旋化。另一方面,催化剂4a和4b都不会促进苄醇和硅烷的脱氢偶联,然而利用钌催化剂5却在14 h后观察到了相当量的硅醚产物,显然催化剂5并不是合适的选择。鉴于催化剂4a和4b在催化活性上没有明显差异,作者后续的实验都是基于4a络合物进行的。
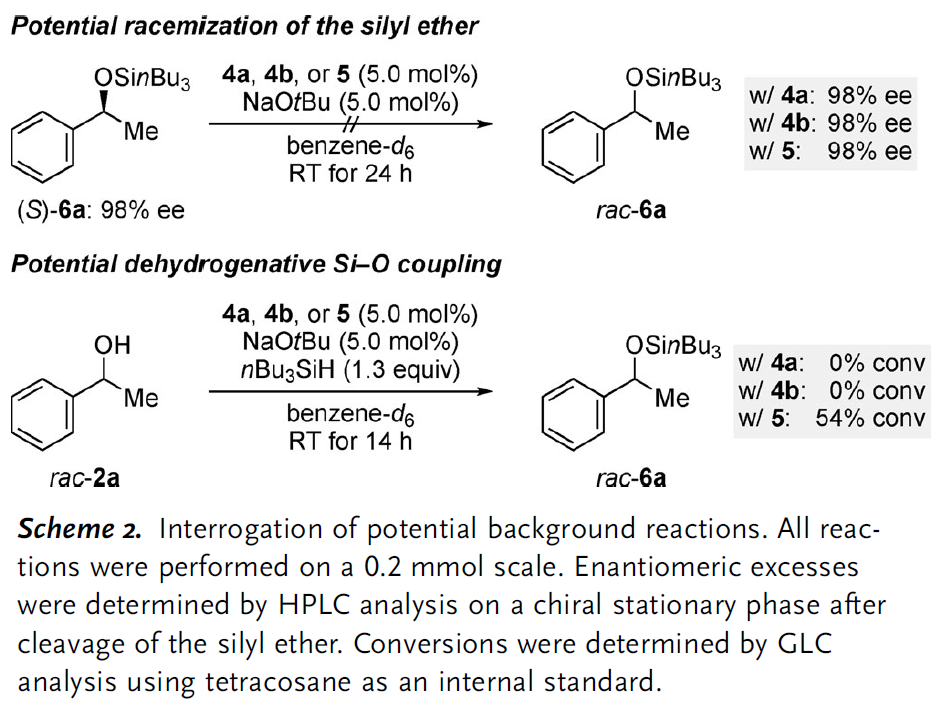
图 3 消旋化催化剂的背景反应测试(图片来源于Angew)
3.底物适用范围
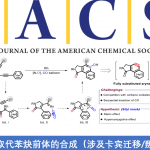
确定最佳的消旋化催化剂后,作者又对模型反应的条件进行了更深入优化并由此建立了标准反应条件,随后他们对反应的底物适用范围进行了探究。多种多样的苄醇都能顺利参与反应,以近乎当量的产率和高对映选择性获得拆分产物(S)-6a-y,并且取代基的电子效应对反应影响不大。对于苯环上的取代基而言,单个甲基取代基无论在邻、间或者对位反应都能发生,仅有ee值微小的差异(2b、2d和2f)。然而,大体积的溴原子在这些位置时对反应的影响较大,特别是位于邻位时的对映选择性特别差(34% ee)。考虑到间位取代基底物能获得更高的ee值,间位双取代的产物都获得了很理想的结果[(S)-6n-p],特别是和邻位双取代的产物(S)-6q相比,由此不难看出邻位的位阻效应对消旋化催化剂4a影响甚大。除苯环外,其他的芳香环体系,例如含萘基(2r和2s)、呋喃基(2t)、噻吩基(2u)、吡啶基(2v)等的底物都能很好的参与该反应。根据作者前期的实验结果,拆分的反应性及对映选择性都会随着烷基基团体积的增大而减小,这一结论在该反应中也得到了比较好的验证,即体积大小:Me < Et < Bn << iPr,产物2a 和2w–y (S)-6a和(S)-6w–y]。需要指出的是,这一趋势在另一个度洛西汀前体底物7中也得到验证,尽管噻吩基底物的兼容性已经得到验证[(S)-6u],但右侧支链却给反应带来很大影响(62% ee)。另一个有价值的产物是含有氨基甲酸酯结构的卡巴拉汀前体[(S)-10,94% yield,93% ee]。

图 4 非环状苄醇的DKR底物适用范围(图片来源于Angew)
作者对脂肪族醇类化合物的适用性也进行了一定探究,但没能获得特别理想的结果,许多底物甚至不能完全转化并且这些底物的反应时间差异也巨大(数小时到数日不等),代表性的例子如下图所示。
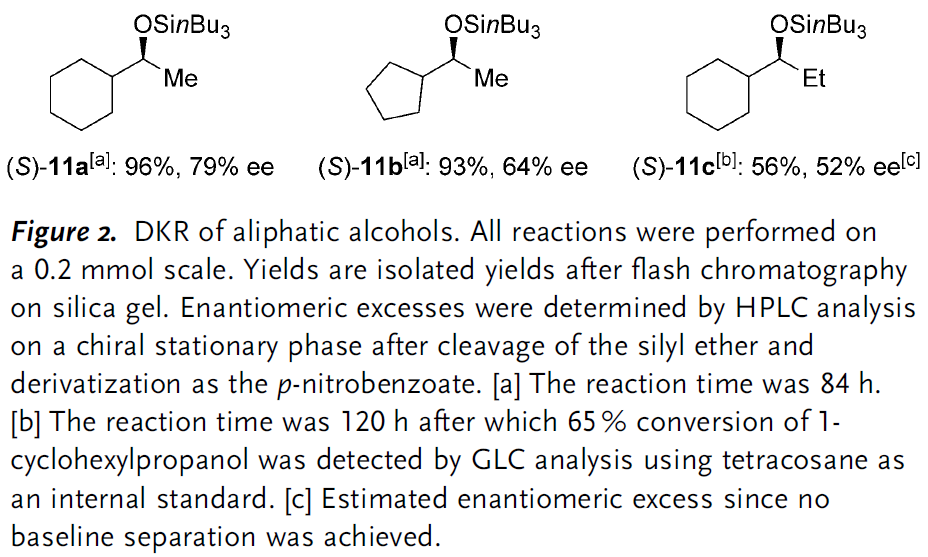
图 5 代表性的脂肪族底物适用范围(图片来源于Angew)
最后,作者还尝试对环状苄醇底物的适用范围进行探究。同样地,这些底物都能获得近乎当量的产率并且对映选择性也较非环状底物略好。具体说来,环的大小对反应影响不大,尽管七元环状化合物(S)-13c的ee值较低。饱和脂肪环中插入O,N,S杂原子后也能很好地兼容,产率和对映选择性都十分可观。
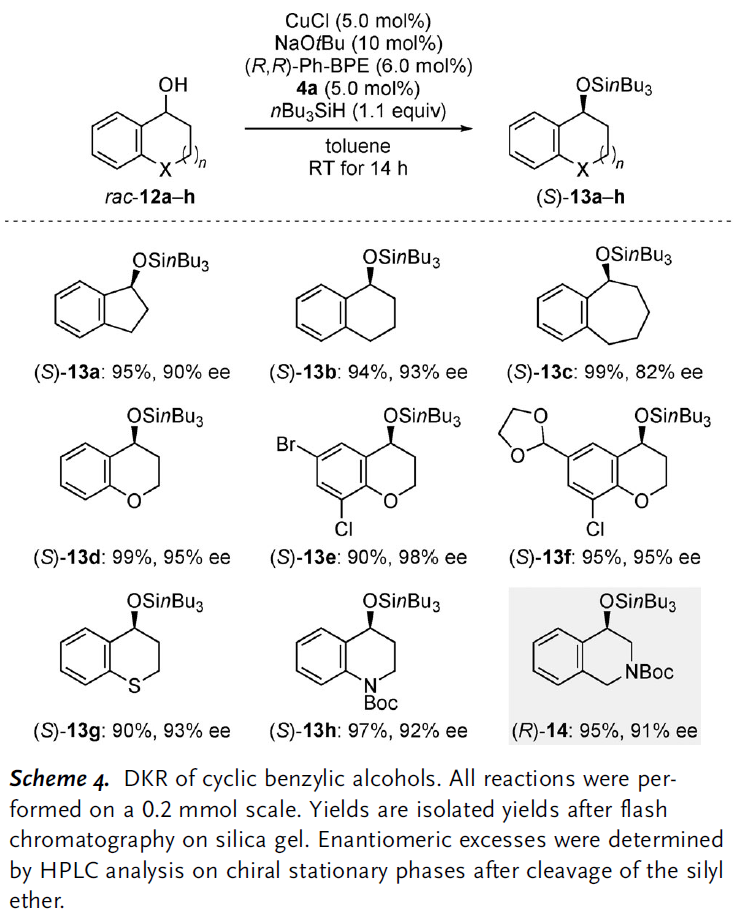
图 6 环状苄醇的DKR底物适用范围(图片来源于Angew)
总结与评价
仲醇的动态动力学拆分通常借助化学酶促的对映选择性酰基化反应实现,Fu等人的非酶促方法不矢为一种“孤立”且漂亮的正交双催化体系。然而,与酰基化同源的对映选择性硅基化方法目前却前所未有。鉴于此,柏林工业大学Martin Oestreich教授等人发展的Cu-H催化醇和简单硅烷nBu3SiH间的对映选择性脱氢偶联反应显得别具一格。该反应体系中,消旋化催化剂的选择及其与拆分体系的兼容性至关重要。该反应具有宽广的底物适用范围及官能团兼容性,也为后续醇类化合物的非酶促DKR过程提供了新的视角和思路。
参考文献
[1] P. M. Dinh, J. A. Howarth, A. R. Hudnott, J. M. J. Williams, W. Harris, Tetrahedron Lett. 1996, 37, 7623-7626. DOI: 10.1016/0040-4039(96)01677-2[2] B. Martín-Matute, M. Edin, K. Bogár, F. B. Kaynak, J.-E. Bäckvall, J. Am. Chem. Soc. 2005, 127, 8817-8825. DOI: 10.1021/ja051576x
[3] S. Y. Lee, J. M. Murphy, A. Ukai, G. C. Fu, J. Am. Chem. Soc. 2012, 134, 15149-15153. DOI: 10.1021/ja307425g





















No comments yet.