

本文作者:杉杉
导读
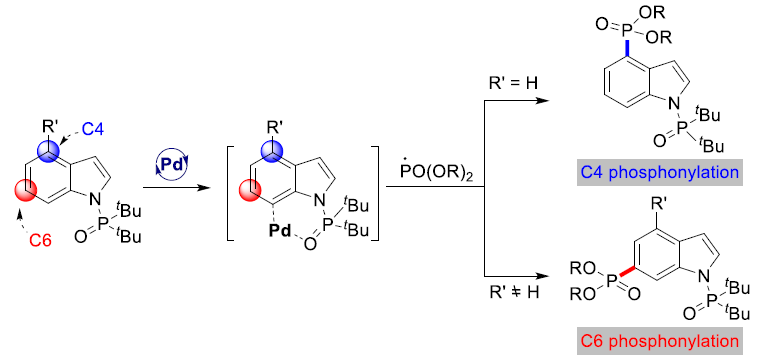
近日,山东大学史大永教授课题组在Angew. Chem. Int. Ed. 上发表论文,报道了一种钯催化吲哚远程C4-H键的膦酰基化反应(phosphonylation),涉及自由基途径,获得了一系列C4-膦酰基化的吲哚衍生物。同时,色氨酸(Trp)和含Trp的二肽也可在C4位置平稳反应(通常依赖C3导向基团的使用)。值得注意的是,使用C4位含有取代的吲哚底物,则可获得C6-膦酰基化吲哚衍生物。初步的机理研究表明,该反应通过C7-钯环化/远程活化过程进行。此外,使用二氟化试剂(BrCF2COOEt)也可进行C4-H二氟甲基化反应。
Palladium-Catalyzed Remote C-H Phosphonylation of Indoles atthe C4 and C6 Positions by a Radical Approach
Xiaolin Shi, Zemin Wang, Yuxiu Li, Xiaowei Li, Xiangqian Li, and Dayong Shi*
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202103395
正文
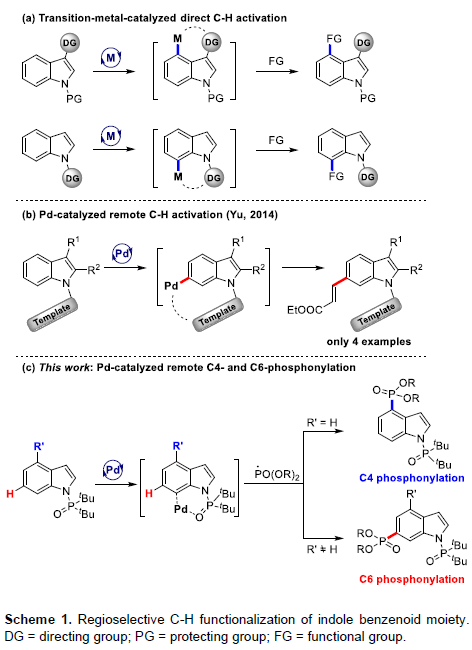
吲哚化合物作为一类重要的含氮杂环骨架,广泛存在于天然生物、药物等中。传统上构建吲哚环主要涉及预官能化吲哚的偶联反应以及新吲哚环的构建,而过渡金属催化吲哚的区域选择性C-H活化则是一种更为高效的策略。然而,吲哚中存在六个独特的C-H键活化位点。其中,虽然吡咯环(C2-C3)具有固有的反应性,但对于苯环(C4-C7)的选择性功能化仍具有挑战。在过去十年中,Shi[1]和Ackermann[2]等,已实现了铱、铑、钌和钯催化吲哚类化合物的直接C-H活化。通常,这些方法使用特定的导向基团在C4或C7位置形成六元金属环,以便后续的氧化加成/还原消除过程(Scheme 1a)。相比之下,Yu等[3]报道了钯催化吲哚的远程C-H功能化,通过导向基团的模板设计,实现了C6烯基化反应(Scheme 1b)。然而,在该方法中需在C2-C3位上具有取代基团的吲哚来保护该部分免于反应。另一方面,磷取代杂环化合物广泛存在于农业、药物化学、生物活性分子等中,但大部分吲哚的膦酰基化反应主要集中于C2和C3位,对于C4-C7膦酰基化反应很少被研究。在此,本文将介绍一种将二叔丁基膦酰基作为有效导向基团(DG),成功实现了钯催化的吲哚的远程C4-H膦酰基化反应。若使用C4取代的吲哚底物,则可实现C6-H膦酰基化反应(Scheme 1c)。同时,此反应涉及C7-钯环/远程活化的过程。
作者以吲哚1a和亚磷酸二乙酯2a作为模型底物,进行了相关膦酰基化反应条件的筛选(Table 1)。反应最佳条件为:Pd(OPiv)2为催化剂,L1为配体,(NH4)2SO4为添加剂,Ag3PO4和K2S2O8为助氧化剂,MeCN为溶剂时,可获得C4膦酰基化产物3a,收率为71%。

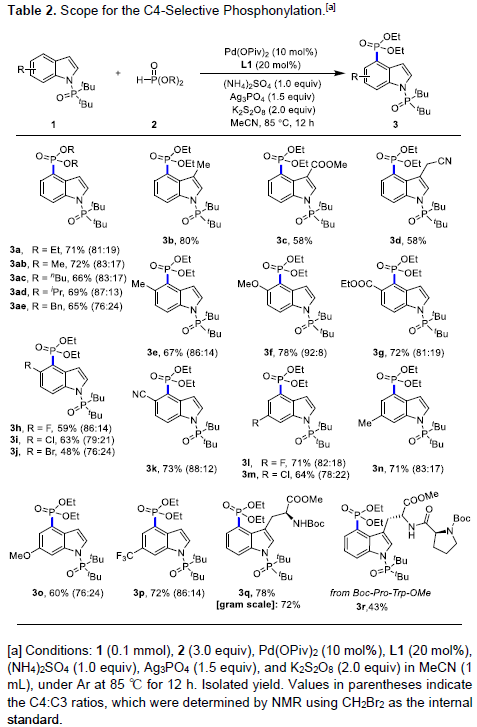
在获得上述最佳反应条件后,作者对C4-膦酰基化的范围进行了扩展(Table 2)。首先,各种取代的磷酸酯2均可与1a顺利反应,获得相应产物3a–3ae,收率为65-72%。其次,具有给电子基、缺电子基和卤素的吲哚底物,均与体系兼容,从而获得相应的产物3b-3p。此外,色氨酸(Trp)和含Trp的二肽也可在C4位顺利反应,获得产物3q(收率78%)和3r(收率43%)。
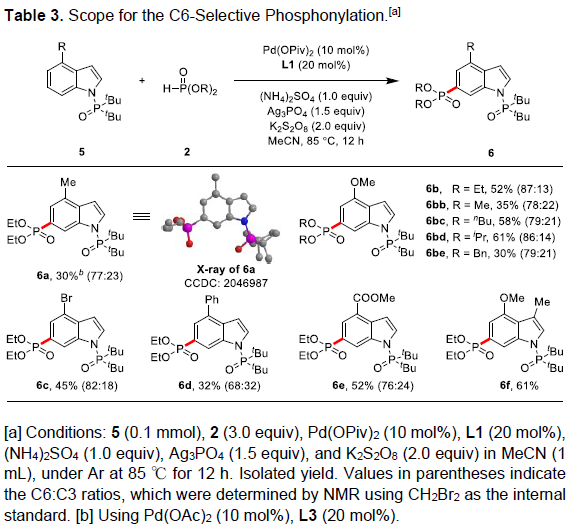
令人兴奋的是,当使用C4位含有取代的吲哚底物时,在标准条件下,则发生C6-H膦酰基化反应。因此,作者对C6-膦酰基化的范围进行了扩展(Table 3)。具有供电子基和缺电子基的底物,均可顺利反应,获得相应产物6a-6f,收率为30-61%。其次,各种取代的磷酸酯2均可与5反应,获得产物6bb–6be。此外,3-甲基-4-甲氧基二取代的吲哚,也与体系兼容,获得产物6f,收率为61%。
为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 2)。首先,在标准条件下加入自由基抑制剂TEMPO或BHT时,未发生反应。而在自由基钟实验中,获得47%收率的化合物7(Scheme 2a-2b)。这两个实验均表明反应涉及自由基的途径。其次,当吲哚的C7位含有甲基和苯基时,在标准条件未能获得C4-膦酰基化产物3s和3t,仅获得未反应的底物,从而表明关键的钯环中间体需在C7位成环(Scheme 2c)。此外,1a和2a在D2O中反应时,C7位未观察到氘的掺入,可能是因为在协同金属化-去质子化过程中,C7中的H原子转移到Lewis碱性膦酰基DG中的O原子上。因此,1a和2a在标准条件下于CH3COOD中反应,获得23%收率的[Dn]-1a和60%收率的[Dn]-3a(Scheme 2d)。氘以大量掺入C7位置,而C2没有氘。同样,[Dn]-1a与2a在标准条件下于水或CH3COOH中反应时,未观察到氘的损失(Scheme 2e)。上述结果表明,反应可能通过C7-环金属化/远程活化途径进行。
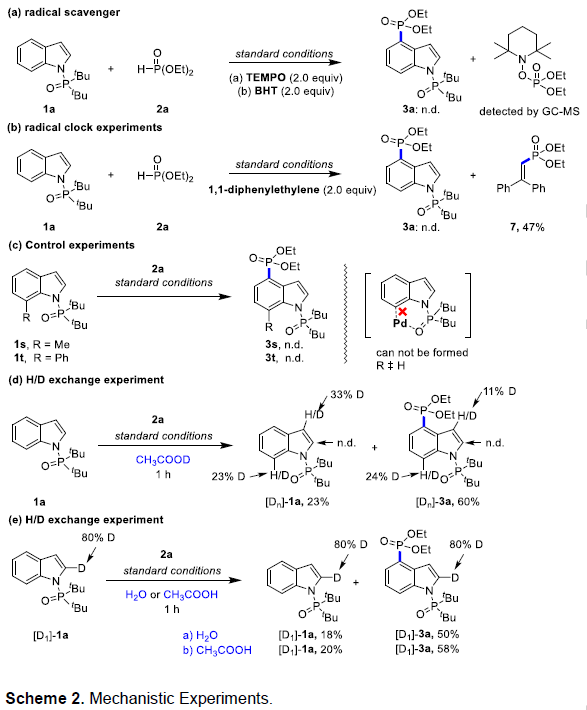
随后,作者提出了一种可能的反应机理(Scheme 3)。首先,钯配合物A与吲哚底物通过环金属化反应以及C7处氢原子转移,形成六元钯环中间体C。然后,亚磷酸二乙酯在氧化剂辅助下,形成膦自由,并对钯环进攻,并且C6-H键在C4位被活化,从而生成中间体D或D’。随后,配合物D或D’与过硫酸根离子发生单电子转移(SET)以及重新芳构化,形成配合物F或F’。最后,通过去质子化即可所需的产物3a或6a,并再生催化活性钯(II)配合物A。
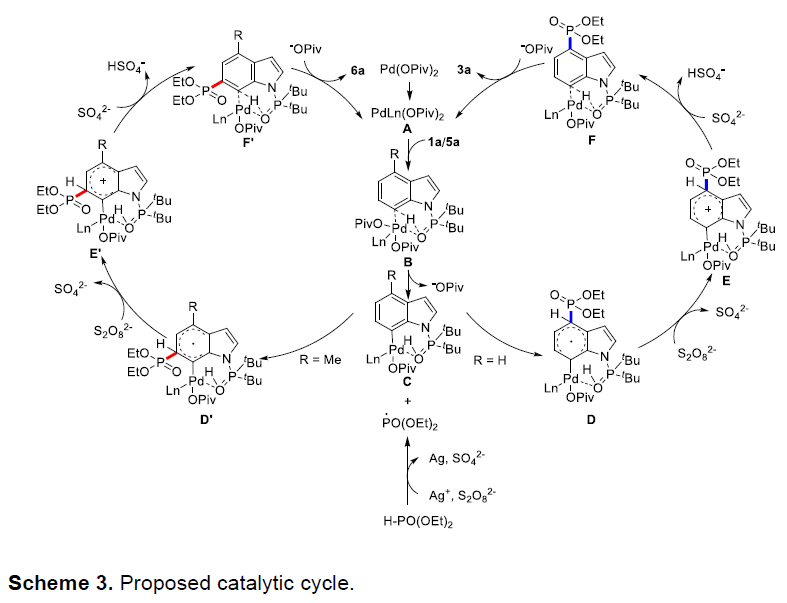
此外,该策略也成功应用于吲哚的二氟甲基化反应(Table 4)。当在PdCl2和Xantphos催化体系中,以BrCF2COOEt(8)为二氟化试剂,可实现C4位二氟甲基化反应,获得产物9a–9c。不幸的是,在优化的标准条件下,对于C4位含有取代的吲哚底物5,未能获得C6位二氟甲基化产物。
总结
本文主要报道了一种钯催化吲哚远程C4-H键的膦酰基化反应。同时,若吲哚的C4位含有其它取代基时,则可实现C6-H键的膦酰基化反应。初步机理研究表明,反应涉及C7-钯环化/远程活化的过程。值得注意的是,导向基团的空间和电子效应对于选择性的控制至关重要。此外,使用二氟化试剂(BrCF2COOEt)也可进行C4-H键的二氟甲基化反应。














No comments yet.