
本文作者:Sunny华
导读
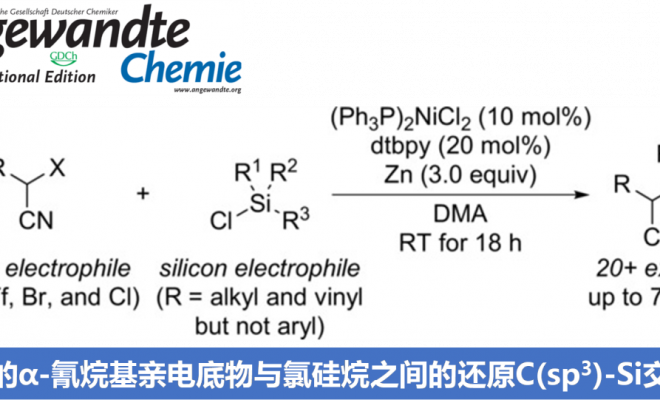
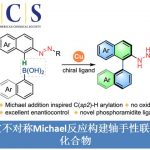
德国柏林工业大学M. Oestreich课题组采用镍/锌催化体系,成功实现α-氰基活化的烷基亲电底物与氯硅烷间的还原交叉偶联反应方法学。其中,金属锌作为化学计量还原剂。通过上述偶联过程,该小组采用两种不同类型的亲电底物,顺利完成C(sp3)-Si键的构建。而之前文献报道的C(sp3)-Si键构建的策略中,则选用碳亲核试剂与硅亲电试剂或碳亲电试剂与硅亲核试剂的组合。
Nickel-Catalyzed Reductive C(sp3)-Si Cross-Coupling of α-Cyano Alkyl Electrophiles and Chlorosilanes
L. Zhang,M. Oestreich, Chem. Int. Ed.2021, ASAP. doi: 10.1002/anie.202107492.
研究背景
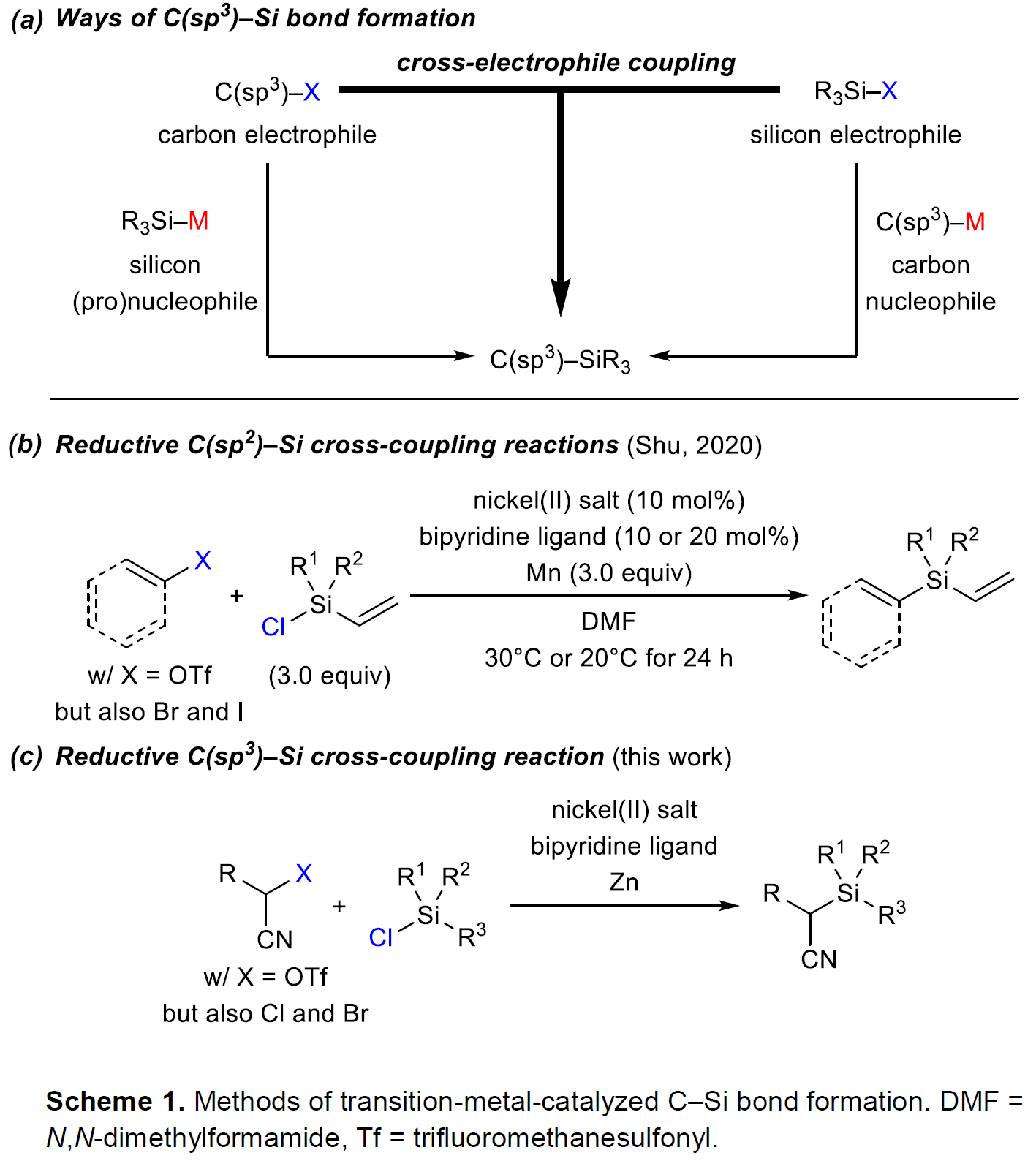
过渡金属催化下,通过碳亲核试剂/硅亲电试剂或碳亲电试剂/硅亲核试剂两种组合方式的交叉偶联过程,进而完成C(sp3)-Si键构建的方法学策略,近年来已经取得较大进展[1]-[5]。然而,该方法学的研究仍存在诸多挑战 (Scheme1, a)。例如,涉及不对称C(sp3)-Si键形成的反应策略[4],目前仅局限于铜催化条件下,活化的烷基亲电底物与带有硼基团的硅前亲核试剂 (boron-based silicon pronucleophile)之间的SN2型反应[4]以及非活化型烷基亲电底物与金属化的硅试剂在无催化剂存在下的取代反应[6]。C(sp3)-Si键构建的另一挑战在于直接将碳亲电试剂与硅亲电试剂应用于还原偶联过程,而无需将相应的偶联参与物进行预先的金属化处理。2020年,兰州大学舒兴中教授课题组报道采用烯基以及芳基三氟甲磺酸酯/卤代物与氯硅烷之间的还原C(sp2)-Si交叉偶联反应方法学 (Scheme1, b)[7],反应过程中需要采用Ni(II)-联吡啶配合物作为催化剂前体,其中,选择金属锰作为化学计量还原剂。尽管该方法具有良好的应用价值,然而,必须选择带有烯基取代的氯硅烷,进而增强底物与Ni催化剂的配位性能。然而,与此相关的形成C(sp3)-Si键的合成策略,目前尚未有相关的文献报道。基于上述研究报道,Oestreich课题组成功开发出通过Ni催化的活化烷基亲电底物与一系列氯硅烷底物之间的交叉偶联反应方法学 (Scheme1, c)。
研究内容
- 1. 反应条件的优化
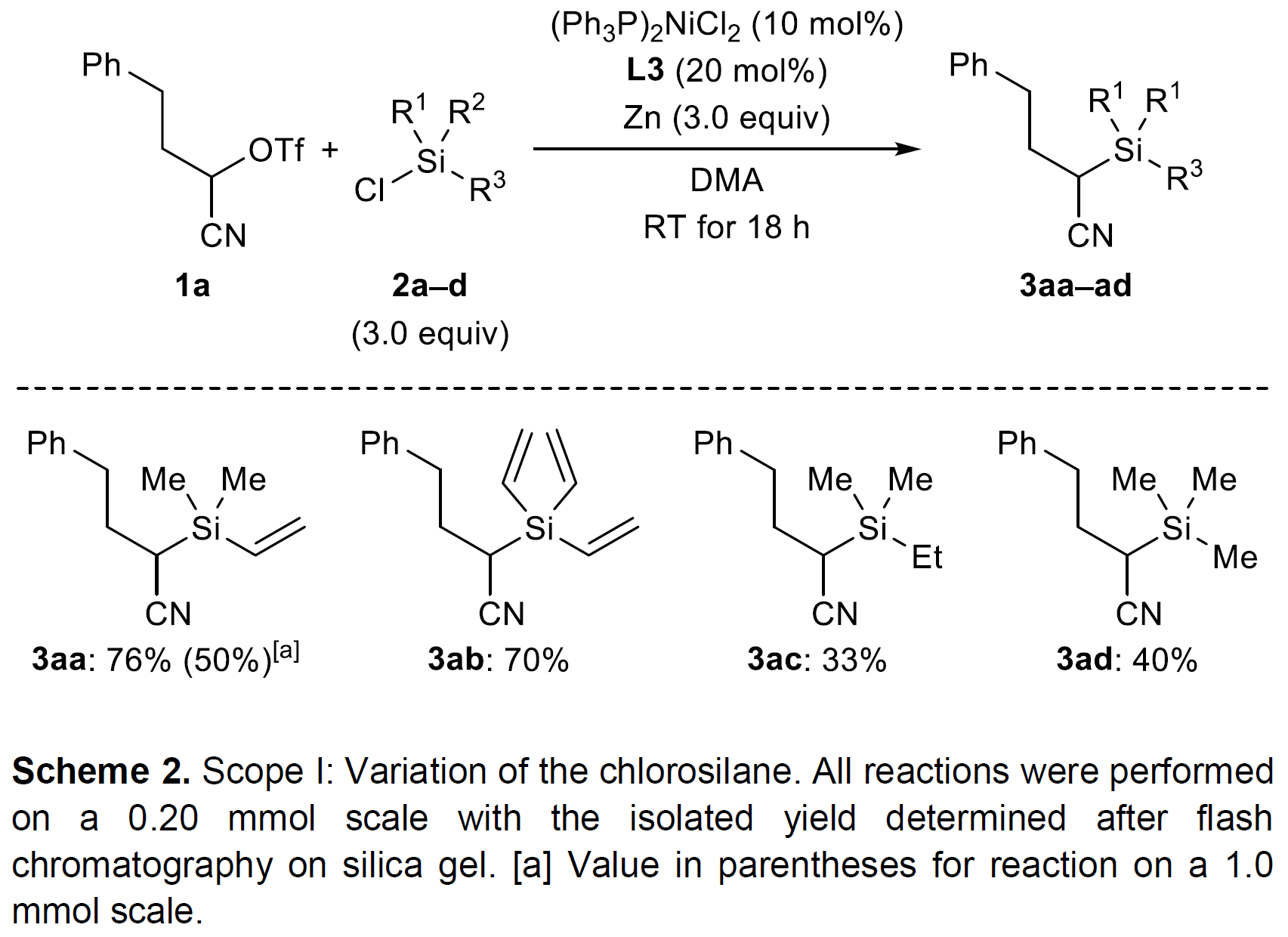
作者采用α-三氟甲磺酰氧基腈 (α- triflyloxy nitrile)1a与乙烯基取代的氯硅烷2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。通过对Ni催化剂、配体、还原剂以及溶剂的进一步筛选,确定最佳的反应条件为:采用(Ph3P)2NiCl2/L3催化体系以及Zn作为还原剂,在N,N-二甲基乙酰胺 (DMA)溶剂中,反应温度为室温,最终以76%的分离产率获得相应目标产物 (entry 1)。同时,通过控制实验的研究表明,化学计量的还原剂锌以及Ni催化剂对于反应的顺利进行尤为重要,并且,研究发现,在无配体加入时,则反应产率较低 (entries 2-4)。并且,选择其他联吡啶配体L1与L2以及三联吡啶L4,目标产物的收率则均低于采用L3作为配体 (entries 5-7),作者进一步发现,采用1,10-邻二氮菲L5作为配体时,能够观察到偶联产物产率急剧降低 (entry 8)。接下来,作者采用金属锰代替锌作为还原剂,同样能够观察到较低的反应收率 (entry 9)。并且,该小组发现采用带有卤素离去基的α-氰烷基亲电底物,同样能够获得良好的反应收率 (entries 10-11)。此外,作者进一步发现,适当地升高或降低反应温度,对于反应收率无显著影响 (entries 12-13)。

- 2. 底物适用范围的考察
在获得最佳的反应条件之后,作者首先考察氯硅烷的底物适用范围 (Scheme 2),研究表明,三乙烯基取代的氯硅烷底物2b能够获得与模型底物2a相近的产物收率。然而,在三烷基取代的氯硅烷2c与2d底物中,尽管缺乏能够与催化剂配位的乙烯基,却同样能够顺利地参与上述偶联过程,并获得中等程度的反应收率 (分别为33%与40%)。这与舒兴中教授报道的实验结果[8]相反,并且,尤其对于通常用作Ni催化还原交叉偶联反应添加剂的Me3SiCl (2d)底物,在上述最佳的反应条件下,同样能够获得相应的C-Si偶联产物[9]。此外,上述反应条件对于硅原子中带有苯基或叔丁基取代的氯硅烷底物而言,则无法获得预期产物,然而,却能够获得相应的二硅烷 (disilane)与二硅氧烷 (disiloxane)产物。
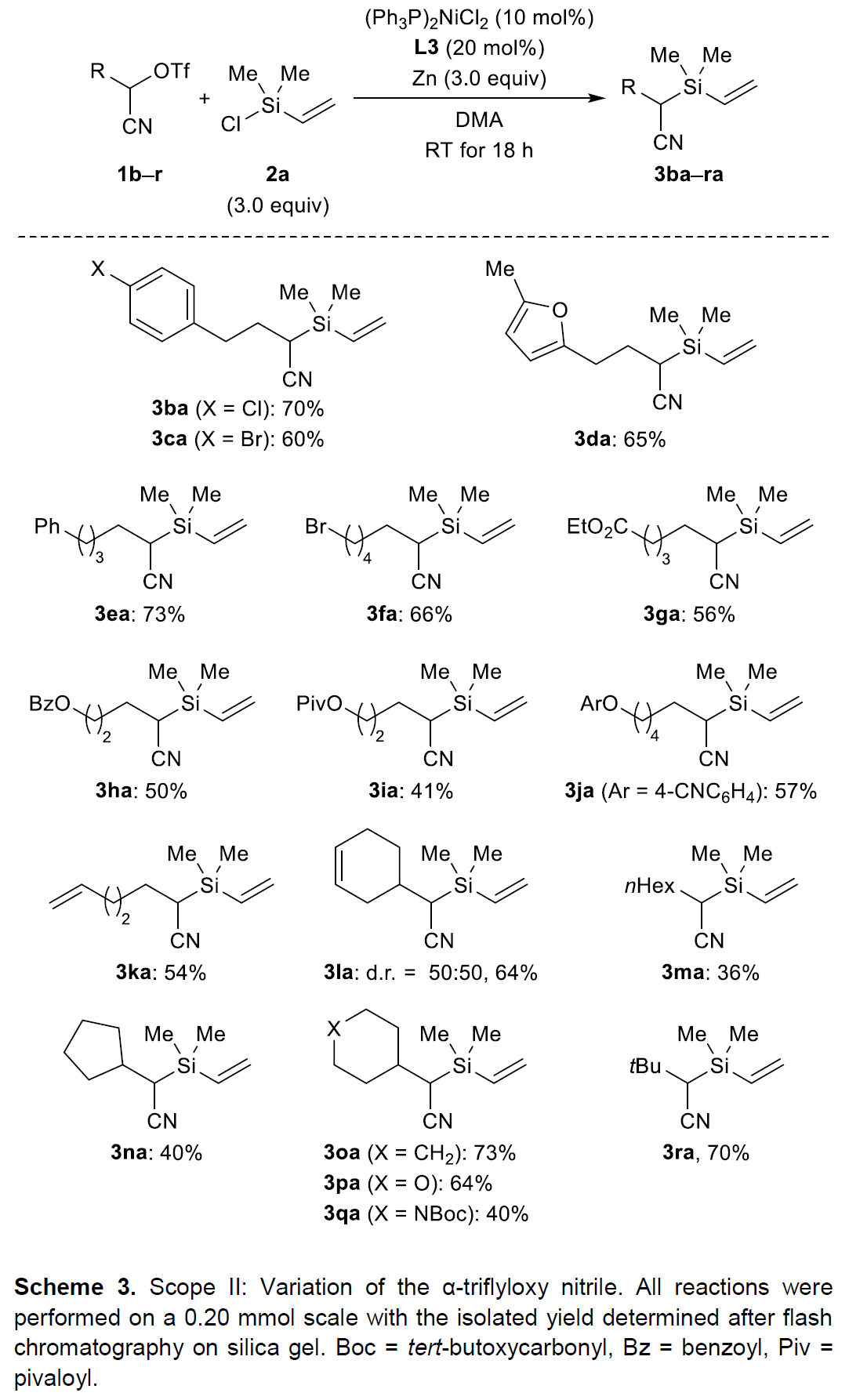
接下来,该小组采用乙烯基氯硅烷2a作为亲电硅底物,对α-三氟甲磺酰氧基腈的底物适用范围进行进一步研究 (Scheme 3)。实验结果表明,芳环中带有卤素取代基的α-三氟甲磺酰氧基腈底物,能够获得中等产率的目标产物3ba与3ca,并且,未观察到芳卤片段参与偶联过程,形成C(sp2)-Si键。此外,上述反应条件对于带有呋喃基取代的α-三氟甲磺酰氧基腈底物,同样能够获得相近收率的目标化合物 (3da,65%)。同时,该小组发现,增加脂肪链的长度,同样能够获得较为理想的实验结果 (3ea,73%)。之后,作者同样对偶联过程中α-三氟甲磺酰氧基腈底物中的官能团兼容性进行深入考察,结果发现,上述反应条件对于一级溴代烷基 (1f)与一系列烷氧羰基官能团取代的α-三氟甲磺酰氧基腈底物 (1g–1j)以及具有不饱和烯键的α-三氟甲磺酰氧基腈底物 (1k与1l)均能够良好地兼容,并获得中等至良好的产物收率。然而,作者进一步发现,上述反应条件对于线性烷基取代的α-三氟甲磺酰氧基腈底物,则仅获得较低的反应收率 (3ma,36%)。然而,对于二级与三级烷基取代的α-三氟甲磺酰氧基腈底物,却能够表现出良好的反应活性 (3na–3ra)。
- 3. 反应机理研究
为阐明合理的反应机理,作者进行一系列相关的控制实验研究。首先,为确定反应过程中是否涉及自由基中间体,作者将过量的TEMPO加入至上述的模型反应体系中,最终,作者观察到,反应收率几乎并未受影响 (Scheme 4,top)。接下来,该小组进一步采用环丙基取代的α-三氟甲磺酰氧基腈底物1s进行相关的自由基探针实验 (radical-probe experiment),结果发现,尽管产物3sa的收率较低,然而,并未检测到开环产物的产生 (Scheme 4,middle)。同时,作者发现,采用相应的α-溴代腈底物,则观察到同样的实验结果,即获得较低收率的3sa (21%)。同样地,在底物1k与2a之间的交叉偶联反应中,并未观察到竞争性的自由基环化过程 (Scheme 3,3ka)。基于上述事实,能够排除反应通过自由基中间体进行的可能性。更为关键的是,该小组选择99% ee的手性纯底物(R)-1a在上述标准条件下进行反应时,则能够观察到彻底的外消旋化 (bottom),进而表明上述的还原偶联过程并非立体专一性反应[10]。

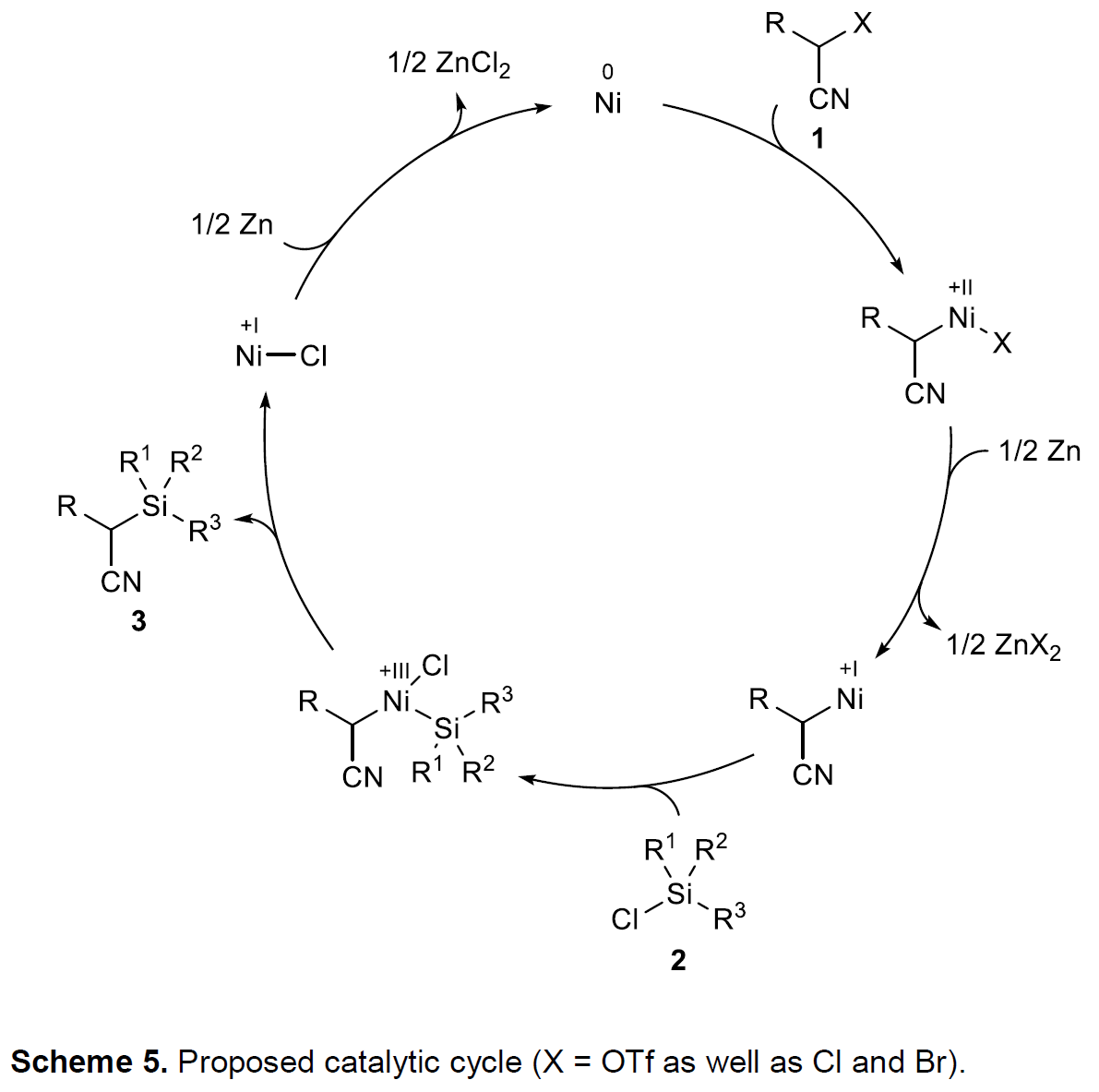
- 4. 反应机理
基于上述实验结果与前期文献报道[11],作者提出一种涉及Ni(0)→Ni(II)→Ni(I)→Ni(III)→Ni(I)→Ni(0)催化循环的机理方案 (Scheme 5)。首先,底物1的C(sp3)-X ( X = OTf, Cl, Br)键与原位形成的Ni(0)配合物通过氧化加成过程,形成烷基Ni(II)中间体,这一机理步骤中,底物将进一步发生外消旋化,尽管缺乏自由基中间体存在的直接证据。接下来,Ni(II)中间体通过Zn的单电子还原过程,获得烷基Ni(I)中间体,该中间体与氯硅烷进行后续的Si-Cl键氧化加成过程,形成烷基(硅基)Ni(III)中间体,之后,经历还原消除步骤,获得相应具有C(sp3)-Si键的目标产物,同时,产生的Ni(I)配合物能够通过Zn的进一步还原,使Ni(0) 配合物再生,进而参与后续的催化循环过程。需要特别指出的是,另一种可能的反应路径,即氯硅烷2的氧化加成先于活化型烷基三氟甲磺酸酯1的氧化加成,目前仍无法排除。
总结与评价
柏林工业大学M. Oestreich教授课题组报道了采用Ni催化剂促进的α-氰烷基亲电底物与氯硅烷之间的还原交叉偶联反应方法学,并顺利实现C(sp3)-Si键的构建。该方法学为首例sp3-杂化的碳亲电试剂与硅亲电试剂之间的还原交叉偶联反应。同时,通过这一策略,能够有效地构建一系列α-硅基腈类化合物。并且,α-硅基腈类化合物能够进一步用于Hiyama交叉偶联反应。此外,将这一全新的偶联策略进一步应用于非活化的烷基亲电底物,并实现不对称催化方式的还原交叉偶联过程,目前仍有待进一步研究。
参考文献
[1] S. Bähr, W. Xue, M. Oestreich, ACS Catal. 2019, 9, 16. doi: 10.1021/acscatal.8b04230. [2] K. M. Korch, D. A. Watson, Chem. Rev. 2019, 119, 8192. doi: 10.1021/acs.chemrev.8b00628. [3] a) C. K. Chu, Y. Liang, G. C. Fu, J. Am. Chem. Soc. 2016, 138, 6404. doi: 10.1021/jacs.6b03465.b) W. Xue, Z.-W. Qu. S. Grimme, M. Oestreich, Am. Chem. Soc. 2016, 138, 14222.doi:10.1021/jacs.6b09596.c) W. Xue, M. Oestreich, Chem. Int. Ed. 2017, 56, 11649.doi:10.1002/anie.201706611.d) H. Hazrati, M. Oestreich, Lett. 2018, 20, 5367.doi:10.1021/acs.orglett.8b02281.e) W. Xue, R. Shishido, M. Oestreich, Chem. Int. Ed. 2018, 57, 12141.doi:10.1002/anie.201807640.
[4] a) J. Scharfbier, H. Hazrati, E. Irran, M. Oestreich, Org. Lett. 2017, 19, 6562. doi: 10.1021/acs.orglett.7b03279.b) J. Scharfbier, B. M. Gross, M. Oestreich, Chem.Int. Ed. 2020, 59, 1577.doi: 10.1002/anie.201912490.
[5] a) M. Takeda, R. Shintani, T. Hayashi, J. Org. Chem. 2013, 78, 5007. doi: 10.1021/jo400888b.b) Z. Huang, R. Ding, P. Wang, Y. Xu, T. Loh, Commun. 2016, 52, 5609.doi:10.1039/C6CC00713A.c) C. Zarate, R. Martin, Am. Chem. Soc. 2014, 136, 2236.doi:10.1021/ja412107b.d) C. Zarate, M. Nakajima, R. Martin, Am. Chem. Soc. 2017, 139, 1191.doi:10.1021/jacs.6b10998.e) B. Cui, S. Jia, E. Tokunaga, N. Shibata, Commun. 2018, 9, 4393.doi:10.1038/s41467-018-06830-w.f) X. Liu, C, Zarate, R. Martin, Chem. Int. Ed. 2019, 58, 2064.doi:10.1002/anie.201813294.
[6] S. Mallick, E.-U. Würthwein, A. Studer, Org. Lett. 2020, 22, 6568. doi: 10.1021/acs.orglett.0c02337. [7] J. Duan, K. Wang, G. Xu, S. Kang, L. Qi, X. Liu, X. Shu, Angew. Chem. Int. Ed. 2020, 59, 23083. doi: 10.1002/anie.202010737. [8] J. Duan, K. Wang, G.-L. Xu, S. Kang, L. Qi, X.-Y. Liu, X.-Z. Shu, Angew. Chem. Int. Ed. 2020, 59, 23083. doi: 10.1002/anie.202010737. [9] N. T. Kadunce, S. E. Reisman, J. Am. Chem. Soc.2015, 137, 10480. doi: 10.1021/jacs.5b06466. [10] J. Scharfbier, H. Hazrati, E. Irran, M. Oestreich, Org. Lett. 2017, 19, 6562. doi: 10.1021/acs.orglett.7b03279 [11] a) A. H. Cherney, N. T. Kadunce, S. E. Reisman, J. Am. Chem. Soc. 2013, 135, 7442. doi: 10.1021/ja402922w.b) X.-G. Jia, P. Guo, J. Duan, X. Shu, Sci. 2018, 9, 640.doi: 10.1039/C7SC03140H .



















No comments yet.