
导读
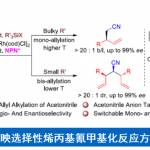
德国柏林工业大学Martin Oestreich教授(主页)等人利用主族B(C6F5)3催化剂,实现了非环状三级胺和二氢硅烷的连续β,β’-选择性硅基化反应,进而发展了高效构建4-硅杂哌啶衍生物的新方法。机理研究表明,反应涉及底物分子中两个烷基部分“胺到烯胺”的脱氢以及烯胺的分子间和分子内两步亲电硅基化过程。日前,相关成果以“热点论文(Hot Paper)”形式在线发表于著名高水平化学期刊——德国应用化学。
Consecutive β,β’-Selective C(sp3)-H Silylation of Tertiary Amines with Dihydrosilanes Catalyzed by B(C6F5)3
Huaquan Fang, Kaixue Xie, Sebastian Kemper, and Martin Oestreich*
Angew. Chem. Int. Ed. 2021, ASAP. DOI: 10.1002/anie.202016664
研究背景
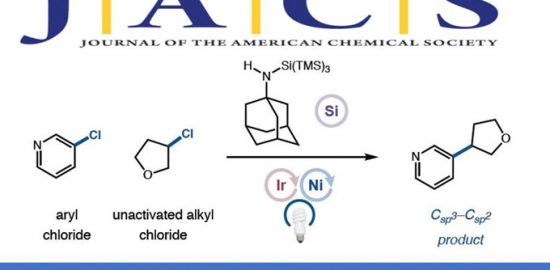
C(sp3) –H键的选择性官能团化反应是合成化学的重要目标,过渡金属催化的C(sp3) –H硅基化反应是实现上述目标的手段之一,最近含硼Lewis酸也可以用来催化这类反应。例如,B(C6F5)3可以从胺的α-C(sp3) –H键攫取氢负离子从而生成亚胺离子和硼氢化物;亚胺离子随即可以去质子生成烯胺产物(Scheme 1,a)[1]。另一类反应是B(C6F5)3催化胺的β-官能化反应,这些反应首先经历脱氢形成烯胺的过程,然后烯胺β-位进攻亲电试剂实现形式上的β-C(sp3) –H活化,目前此策略已成功应用于环状及非环状三级胺ꞵ-位的硅基化、炔基化、氘代和烯基化等反应(Scheme 1,b)[2]。然而,就硅基化反应而言,Park和Chang课题组虽然以N-芳基哌啶(环状三级胺)为起始原料通过B(C6F5)3催化分子内Friedel-Crafts类型的硅基化反应实现了三级胺的β-C(sp3) –H官能化以及桥环含硅氮杂环化合物的构建[2a],然而非环状三级胺的β-C(sp3) –H硅基化以及更具挑战性的两次C(sp3) –H硅基化反应目前却尚未可知。
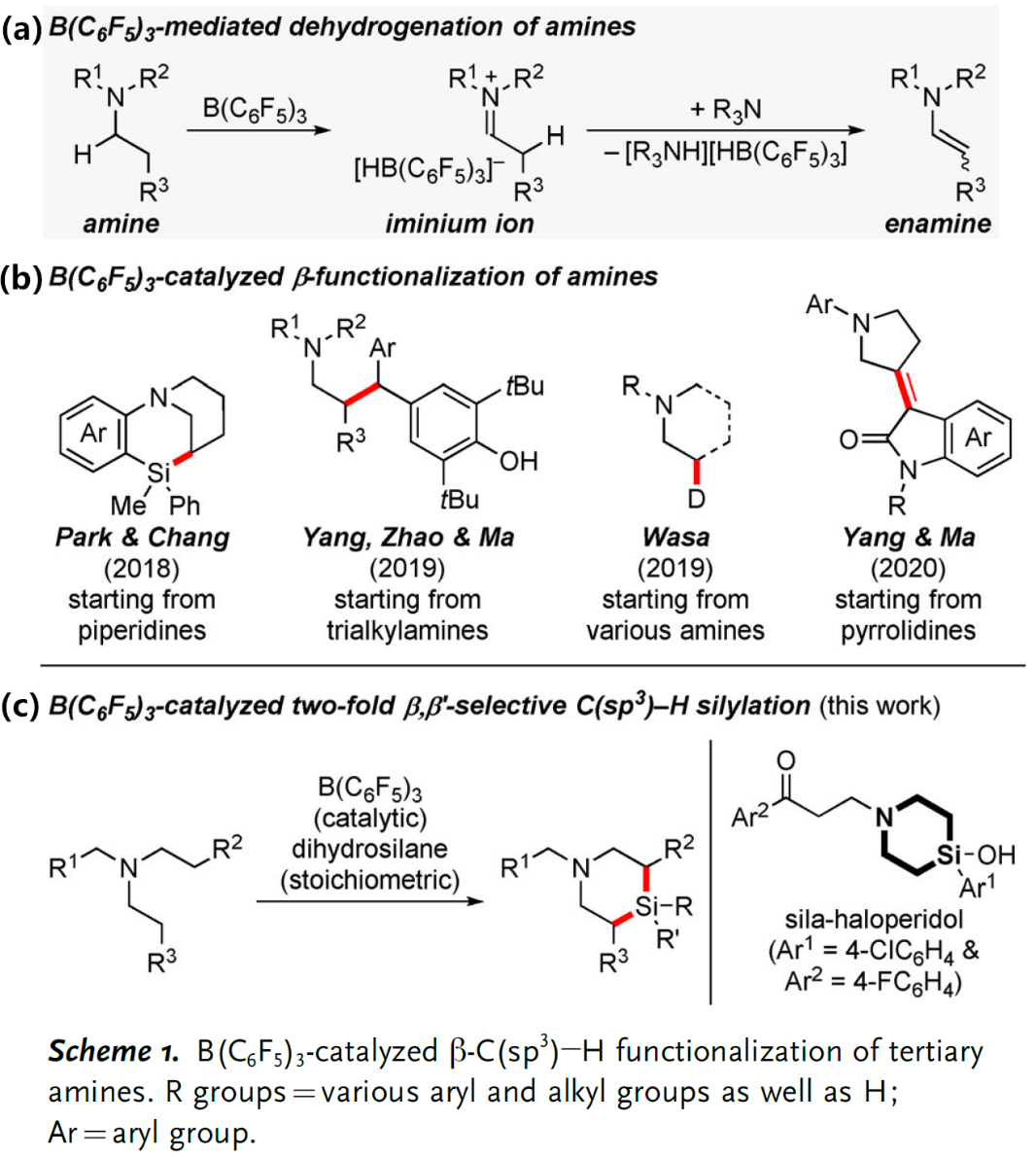
鉴于此,Oestreich课题组另辟蹊径发展了非环状三级胺在B(C6F5)3催化下与二氢硅烷的连续β,β’-选择性C(sp3) –H硅基化反应,实现了硅杂哌啶化合物的快速合成(Scheme 1,c);这类结构是药物化学中有价值的“合成砌块”,例如,多巴胺受体拮抗剂sila-haloperidol中就含有这类硅杂哌啶单元。然而,目前合成这类化合物的方法通常是从双烯基取代硅烷出发的多步骤合成方式,因此上述新方法具有无法比拟的优势。
研究内容
(1)反应条件优化
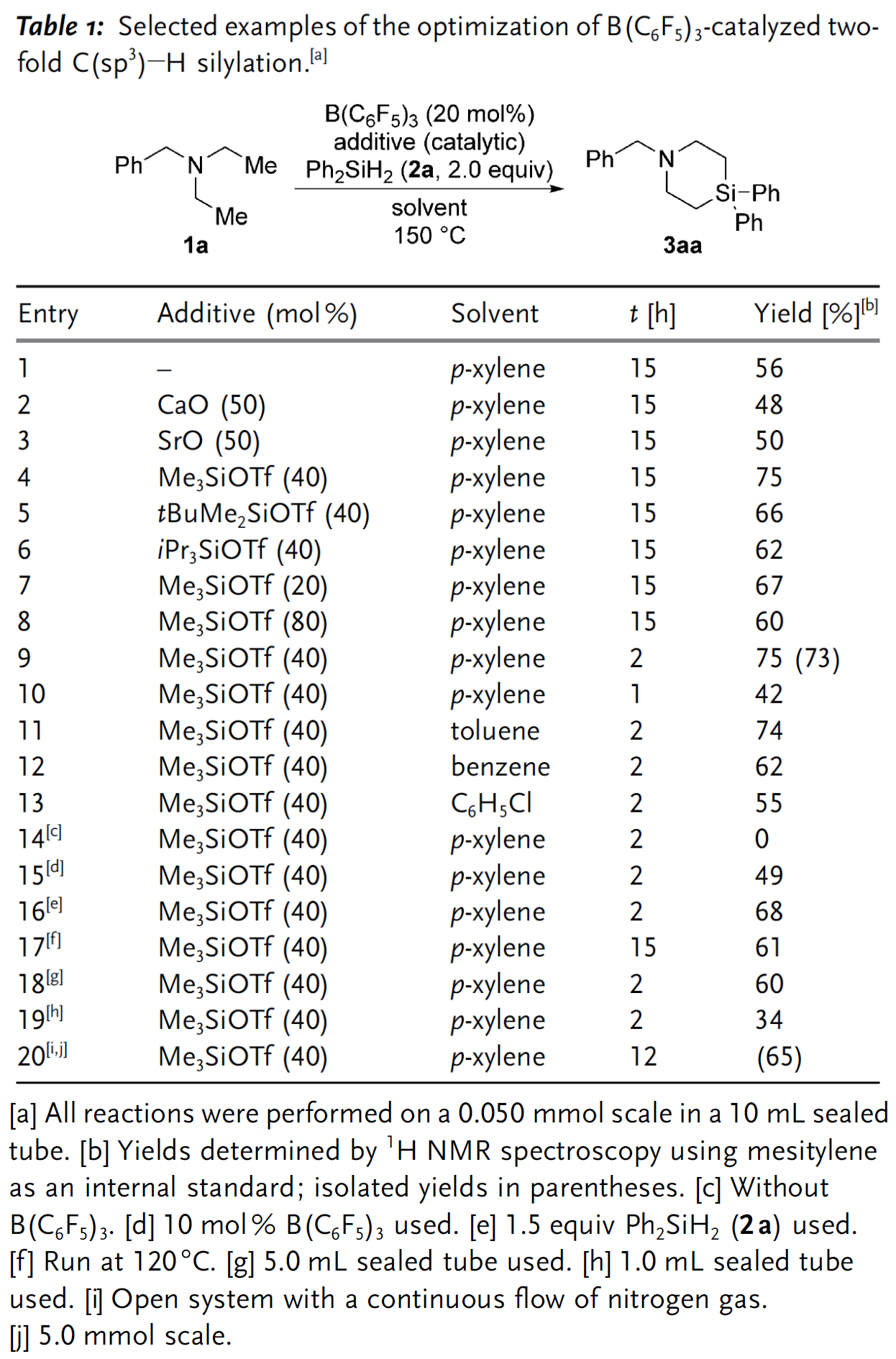
作者首先以二乙基苄胺1a和二苯基二氢硅烷(Ph2SiH2)2a为模型底物对反应条件进行筛选优化。当加入20 mol%的B(C6F5)3催化剂,在对二甲苯中以150 oC反应15 h后可获得中等产率的目标产物3aa(Table 1,entry 1)。根据文献报道,金属氧化物和硅基三氟甲磺酸酯作为添加剂时可以提高该反应活性,然而当作者加入CaO和SrO后产率反而降低(Table 1,entries 2-3),仅在加入40 mol%的Me3SiOTf后反应产率得到明显提升(Table 1,entries 4-6),并且Me3SiOTf的用量过高过低都不利于反应的进行(Table 1,entries 7-8)。随后,作者又针对反应时间、溶剂、温度、反应容器的尺寸等因素进行了细致优化,最终获得最佳的反应条件(Table 1,entry 9)。
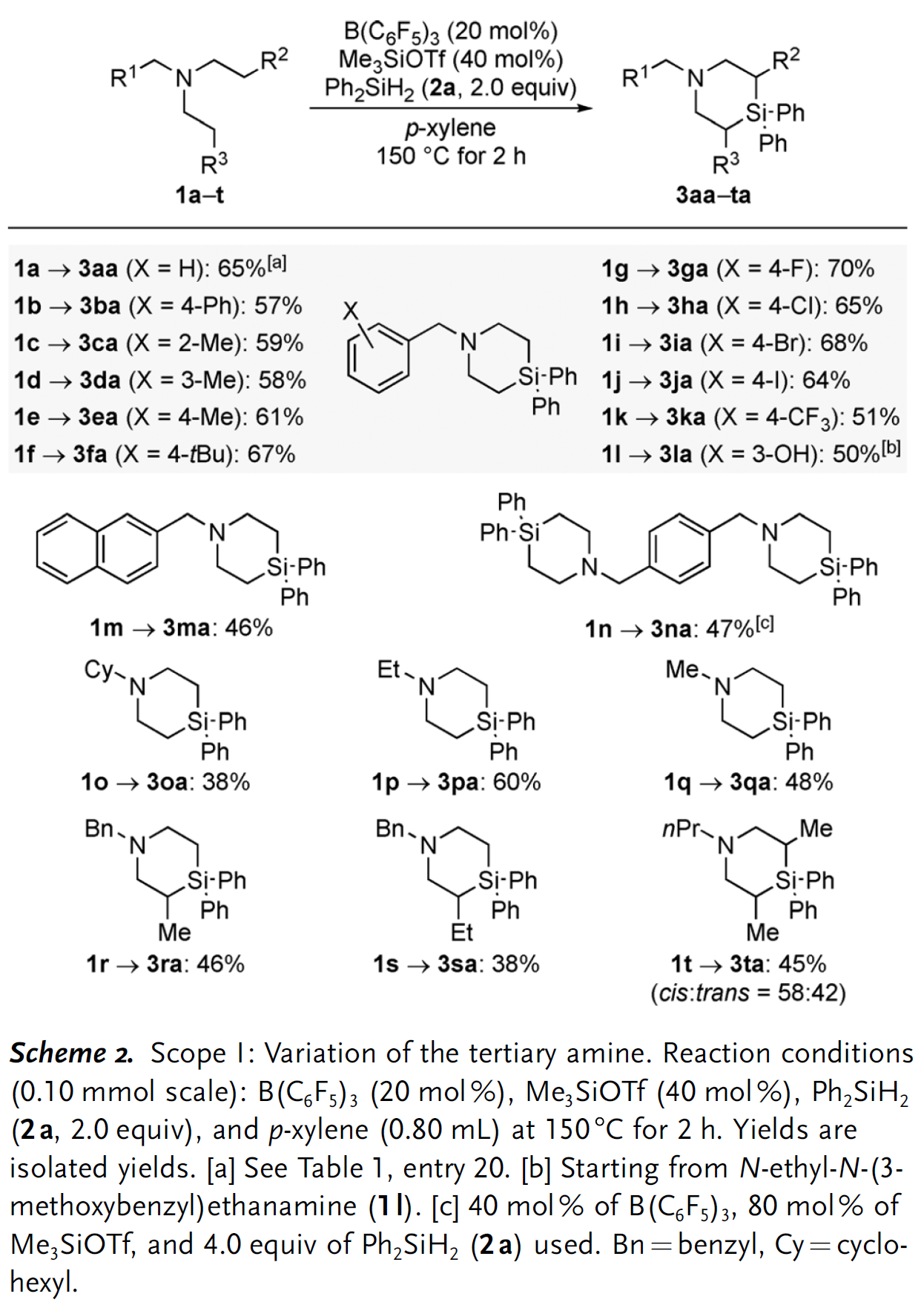
(2)底物适用范围
当获得最佳反应条件后,作者开始对底物的适用范围进行探究(Scheme 2)。需要说明的是,C(sp3) –N键的还原断裂是该反应普遍存在的竞争反应,最终导致生成二级胺副产物[3]。N-苄基二乙胺衍生物的芳环上带有不同电性效应的取代基均能和Ph2SiH2反应生成对应的4-硅杂哌啶产物,并且产率中等偏上(3ba–3la);此外,所有的卤素原子(3ga–3ja)和三氟甲基官能团(3ka)都能兼容该反应。有意思的是,含有甲醚官能团的底物在该反应条件下会发生脱甲基/硅基化过程,最终以50%的中等产率分离得到含酚羟基的产物3la。当苄基的苯环替换成萘基时,产率降低比较明显(3ma)。值得一提的是,如果底物中含有两个三级胺单元,最终可以获得通过四次C(sp3) –H硅基化过程获得对称的双硅杂哌啶产物3na。底物三级胺氮原子上的取代基不局限于苄基,其他烷基取代基(1p和1q),特别是环己基取代的底物(1o)也能选择性在乙基部分发生硅基化生成产物3oa。此外,当分子中的某个乙基替换成丙基或丁基时,反应均能顺利发生(3ra和3sa),而两个乙基均替换成丙基时反应可能产生非对映异构体,实际获得的产物并不具备显著的非对映选择性,顺反异构体比例仅为58:42。
随后,作者又对二氢硅烷的底物适用范围进行了探究(Table 2),对称及非对称结构的二芳基取代硅烷均能获得预期产物并且产率无明显差异(3ab–3ae)。然而,大位阻取代的二氢硅烷,例如均三甲苯取代的硅烷(2f)就不能参与上述反应;双芳基取代硅烷其中一个芳基替换成甲基或者均替换成烷基时,反应虽然都能发生但产率相对较低(3ag和3ai),但大位阻叔丁基同样也不能发生反应(3aj)。此外,当作者尝试以1-硅杂茚满(2h)为底物合成更具挑战的螺环产物时,仅观察到痕量的目标产物。
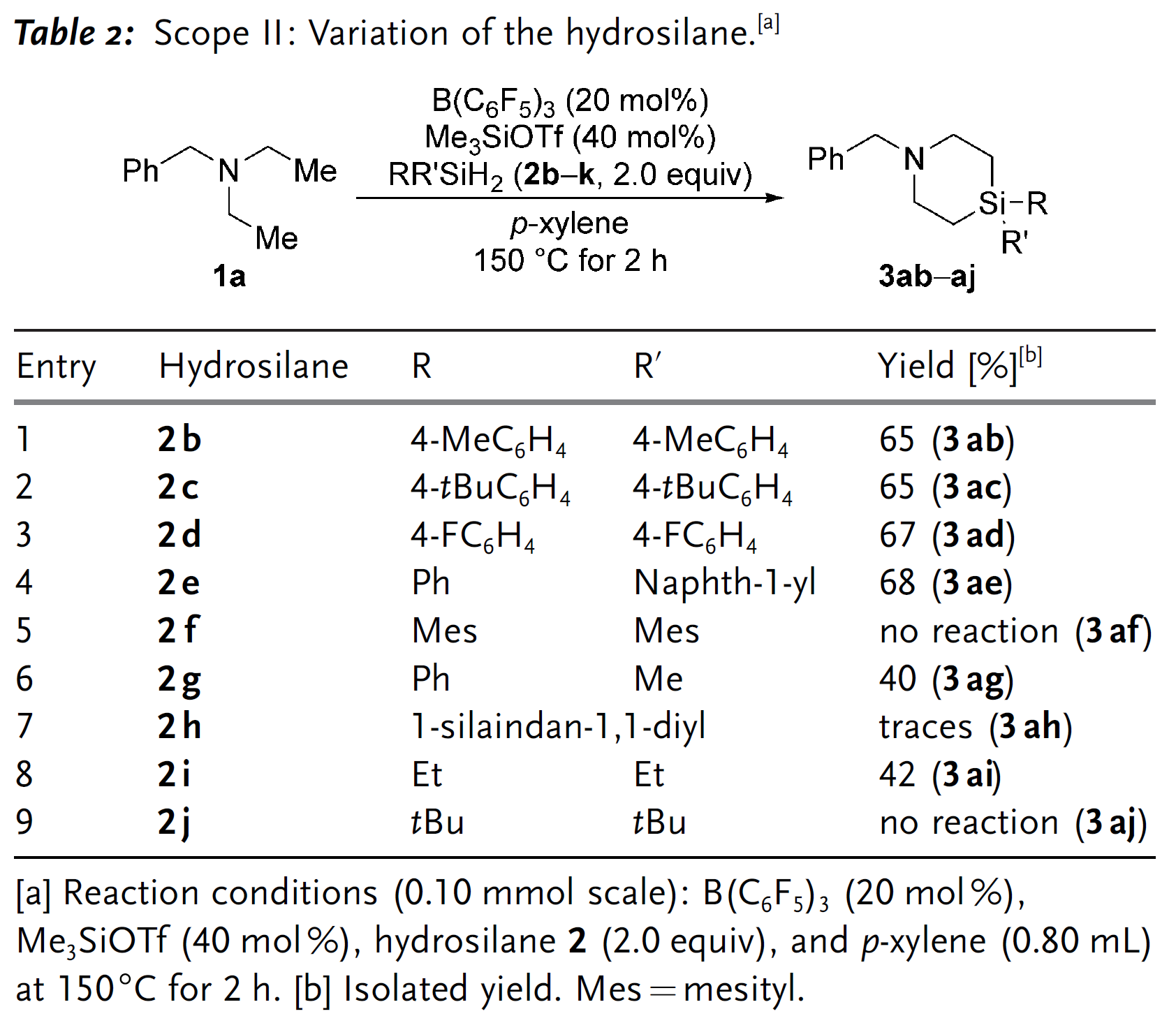
完成底物适用范围探究后,作者又着手对产物进行简单的衍生化(Scheme 4)。产物3aa的苄基可以在氯甲酸-1-氯乙酯(1-chloroethyl chloroformate)和甲醇在加热条件下脱去生成盐酸盐产物6;不仅如此,苄位的C–H键还可以经KMnO4氧化为羰基,最终以中等产率(55%)生成酰胺化合物7。
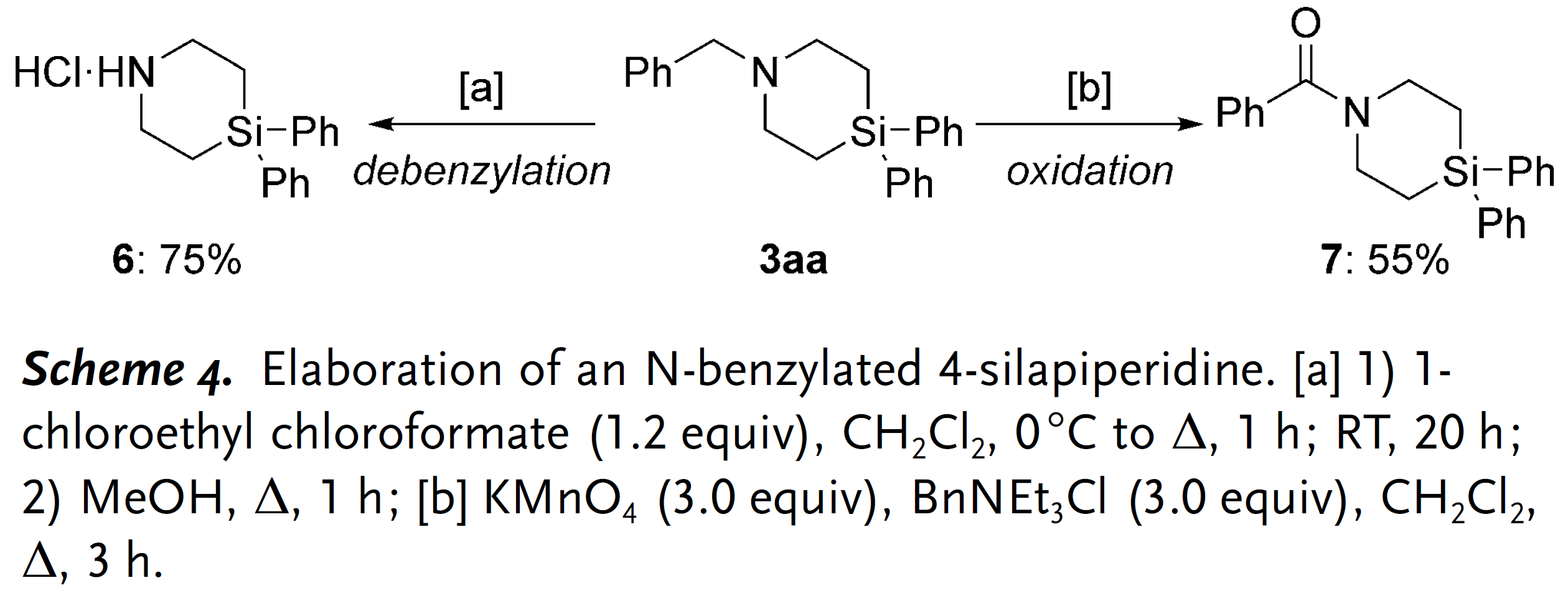
(3)机理研究
为了探究反应的机理,作者设计了相应的控制实验(Scheme 5)。首先,他们进行氘代实验并发现底物1a在标准反应条件下生成的产物3aa共含有88%氘含量,其中苄位和氮原子的α位氘含量均为41%,这意味着氮原子α位C(sp3) –H被攫取氢负离子的过程是可逆的;另外需要注意的是,氮原子的β位也有6%的氘含量,这是烯胺中间体氢化的直接证据。当二乙基取代的1a作为底物时,硅基化生成目标产物的过程速率更快,但二正丁基取代的底物1v在相同反应条件下却并不能得到任何硅基化产物,仅得到氮原子α位氘代后的产物。虽然总氘代率没有变化,但氮原子β位的氘含量却显著提升至24%,这表明烯胺中间体的氢化过程与硅基化过程存在很大竞争。为了探究添加剂Me3SiOTf对反应的影响,作者又进行了一个已知的化学计量实验[1]:将底物1a和B(C6F5)3等摩尔量混合后发现无论是否存在Me3SiOTf,反应的产物种类以及比例都没有发生变化。

(4)反应机理
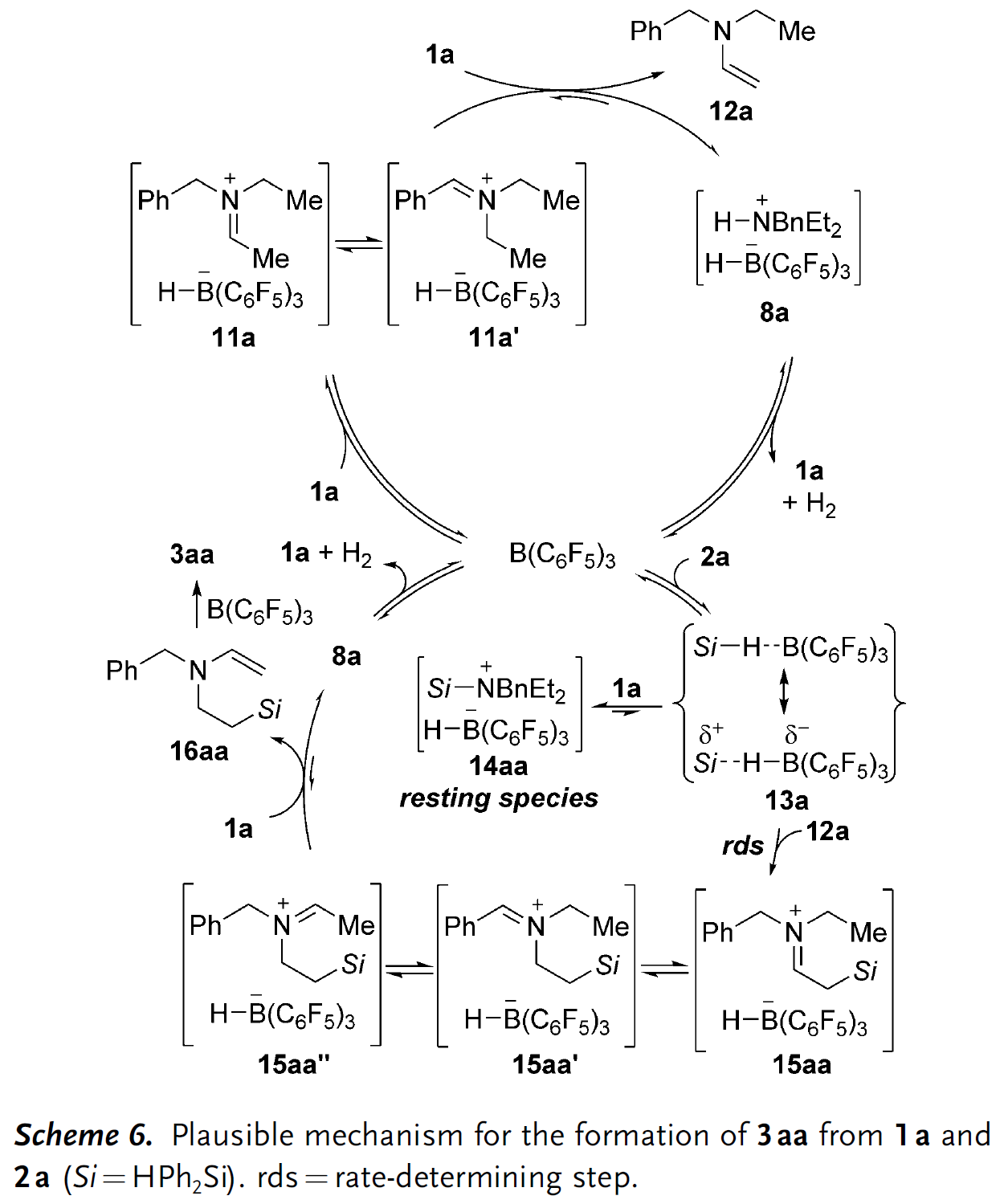
结合上述控制实验的结果以及前期Park和Dang等人辅以DFT计算提出的机理,此处作者也提出了一个可能的反应机理(Scheme 6)。首先,B(C6F5)3从底物1a中攫取氢负离子生成亚胺离子中间体11a及它的平衡异构体11a‘,随后该中间体被未参与反应的三级胺1a去质子得到烯胺中间体12a和FLP(受阻Lewis酸碱对)类型的络合物8a,后者释放出H2后催化剂和三级胺分子可以再生。此外,B(C6F5)3可以活化硅烷2a产生硅亲电试剂,随即被烯胺中间体12a的ꞵ-碳端捕获生成亚胺离子15aa,它存在多种区域互变异构体(15aa‘和15aa”),烯胺亲核进攻硅亲电物种的过程也是整个反应的速率决定步骤。需要说明的是,B(C6F5)3活化的硅烷也可以接受亲核氮原子的进攻生成络合物14aa,它存在于整个反应过程。最初生成的亚胺离子15aa‘经三级胺去质子后得到另一个烯胺中间体16aa并参与B(C6F5)3促进的分子内硅基化过程,最终生成目标产物3aa。
总结与评价
柏林工业大学Martin Oestreich教授课题组报道了一例新颖的B(C6F5)3催化三级胺和二氢硅烷连续β,β’-选择性C(sp3)–H硅基化反应,实现了4-硅杂哌啶衍生物的快速构建。反应涉及底物分子中两个烷基部分“胺到烯胺”的脱氢以及烯胺的分子间和分子内两步亲电硅基化过程,克服了前期类似反应无法适用于非环状三级胺以及无法实现两次选择性硅基化过程的局限。








No comments yet.