

本文作者:杉杉
导读
多氟芳烃(Polyfluoroarenes)广泛存在于药物、材料等多个领域。近日,德国马克斯-普朗克研究所Tobias Ritter教授课题组在Angew. Chem. Int. Ed.上发表论文,首次报道了通过光氧化还原脱羧策略,成功实现脂肪族羧酸的多氟芳基化反应(polyfluoroarylation)。同时,该方法具有广泛的底物范围和良好的官能团耐受性。此外,通过对天然产物和药物的分子的后期修饰,进一步证明了该反应的实用性。
Decarboxylative Polyfluoroarylation of Alkylcarboxylic Acids
Tobias Ritter and Xiang Sun
Angew. Chem. Int. Ed. ASAP DOI: 10.1002/anie.202015596
正文
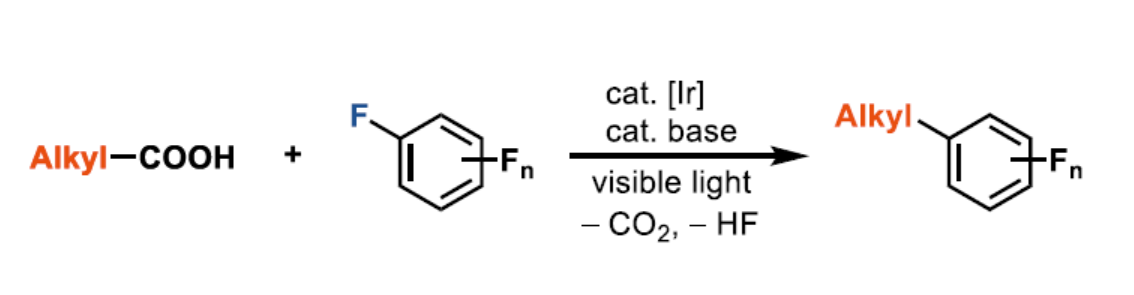
多氟芳烃,由于具有特殊的属性,如低氧化电位和代谢稳定性等,已广泛存在于药物、材料和农药中。通过C-X和C-H键的氟化反应作为合成单氟化芳烃的有效方案,但也可通过多氟芳烃的脱氟官能团化来实现多氟芳烃的合成。同时,氟化物的亲核芳基取代(SNAr)和过渡金属催化的C-F键功能化作为两种主要的途径。然而,在此类反应中,常需预先形成的有机金属试剂,如烷基或芳基锂,-格氏试剂或-锌试剂,从而限制了底物的范围。在此,本文报道了第一个脂肪族羧酸的脱羧多氟芳基化反应,涉及光氧化还原脱羧形成烷基自由基,以及单电子还原生成氟芳基自由基的过程。
通过Halex工艺[1],可轻松合成各种多氟芳烃。对于复杂的小分子而言,通过使用有机金属试剂的脱氟烷基化进行衍生反应具有挑战性。为避免使用化学计量的烷基金属试剂,也可由烯烃催化原位生成烷基铜酸盐,但反应仅限于苯乙烯和活性烯烃[2]。Weaver等[3-4] 通过光催化单电子还原C-F键形成的自由基,从而实现烯烃、炔烃和芳烃的全氟(杂)芳基化反应。尽管具有广泛的底物范围,但为了抑制竞争性的抽氢转移,通常需要过量的烯烃。Hashmi等[5]报道了叔苯胺的多氟芳基化反应,涉及自由基-自由基偶联的过程。在此,德国马克斯-普朗克研究所Tobias Ritter教授课题组报道了在碱和光氧化还原催化剂存在下,从而实现烷基羧酸和多氟芳烃的脱氟芳基化反应,从而获得各种多氟取代的芳烃产物(Scheme 1)。

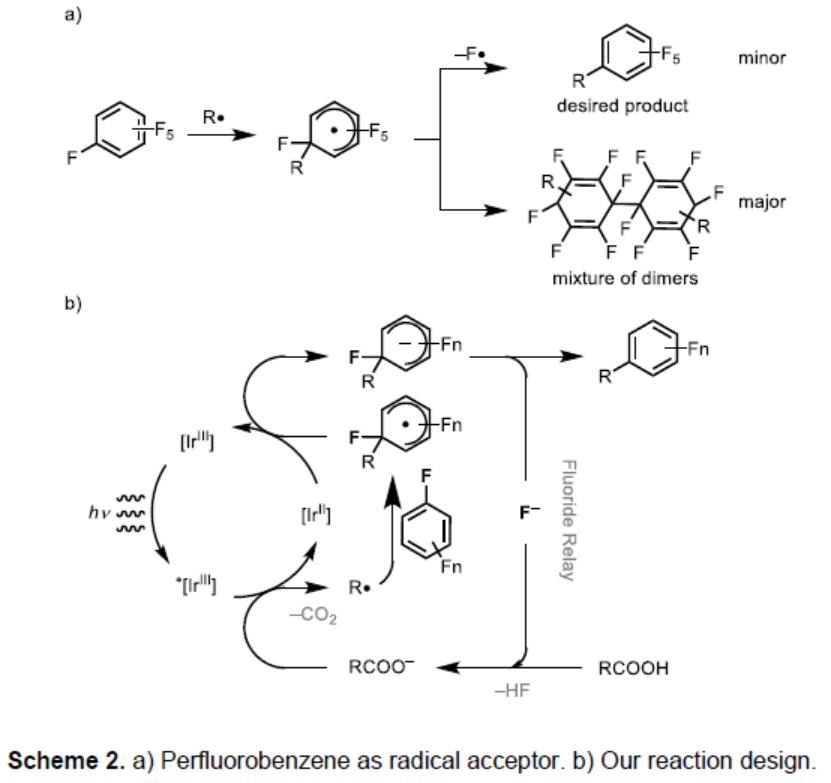
早在1960年,Williams和Kobrina等首次将多氟芳烃作为自由基受体进行反应[6]。由过氧化物生成碳自由基经加成形成烷基化的全氟苯,但也形成自由基二聚化产物(Scheme 2a)。由于C-F键的离解能大,全氟芳烃中自由基加成效率低,并且导致竞争性二聚和加氢烷基化,从而具有较低的合成价值。Weaver等[3,7]报道了全氟芳烃以及缺电子芳烃可通过光氧化还原催化剂还原为相应的自由基阴离子,通过消除氟化物可更易将碳氟键杂化裂解。因此,作者设想,向全氟芳烃中引入一个自由基后,然后将所得的自由基还原为阴离子,从而实现氟化物的消除(Scheme 2b)。
首先,作者对各类烷基羧酸的底物范围进行了扩展(Table 1)。反应结果表明,各种二级脂肪酸(2、3、4和6)、氨基酸(7)、α-氧基取代的羧酸(11和12)等,均可顺利进行反应。同时,对于二羧酸化合物也可实现二全氟芳基化反应(16)。此外,具有羟基(8和10)、羰基(20)、噻唑基(9)、酰胺基(4)、吲哚基(5)等基团时,均可保持构型完整。值得注意的是,对于天然产物(21)和药物分子(13和17)的后期功能化,同样取得预期的结果。

![]()
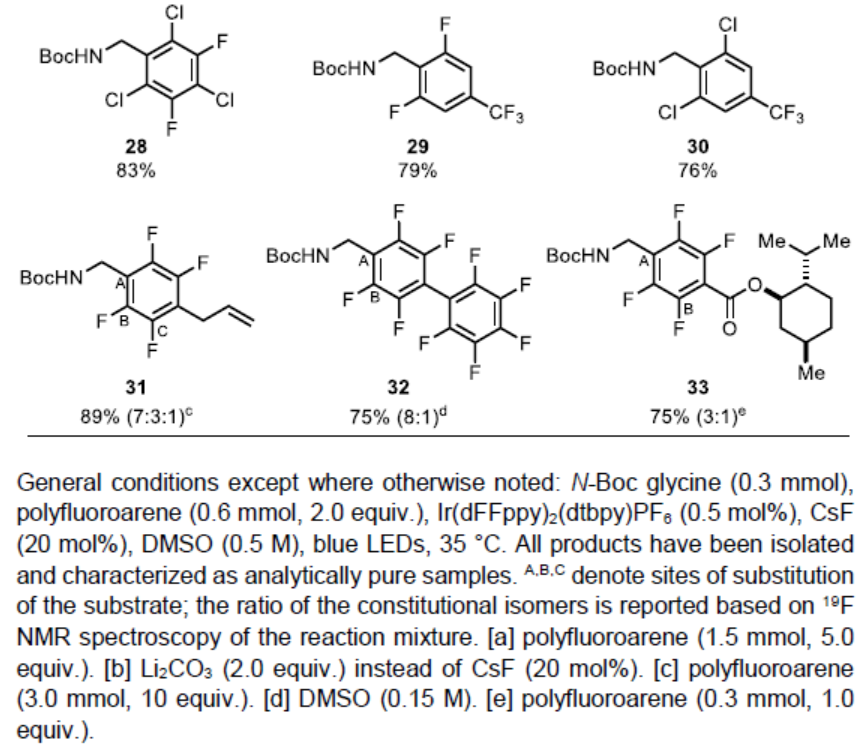
紧接着,作者对多氟芳烃底物进行了相关的扩展(Table 2)。反应结果表明,具有五氟、四氟、三氟、二氟和单氟取代的底物,均可顺利进行反应,获得相应的产物22–33。其中,由于不对称氟代芳烃在不同位置加成而产生的结构异构体,均可通过色谱法纯化(23、24、25、26、31、32和33)。值得注意的是,对于同时含有氟和氯取代基时,反应仅与氟取代反应(23、28和30),与Weaver等报道的反应性相反[3,7]。
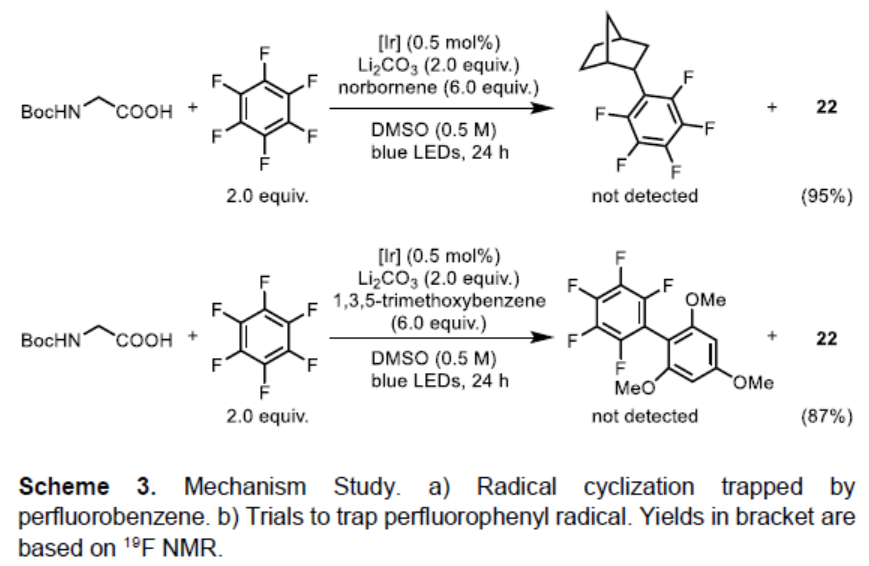
为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 3)。首次,通过自由基环化实验,形成了8%的环化产物34a,同时未观察到目标产物34b,从而表明反应是通过34a来形成碳自由基,在加到氟亚芳基之前5-己烯基自由基可快速经自由基环化(Scheme 3a)。随后,通过还原成阴离子再去除氟化物来再生[IrIII]催化剂即可结束循环。尽管Hashmi课题组[5,8]已提出通过自由基-自由基偶联形成C-C键,但该策略不适合该体系。首先,考虑到氧化还原电势,不太可能发生铱光催化剂对全氟芳烃的单电子还原。此外,通过加入tBu-乙炔,降冰片烯和1,3,5-三甲氧基苯作为清除剂,未观察到全氟苯基自由基加合物(Scheme 3b)。同时,在氯氟芳烃的反应中,观察到的排他性的氟取代,也表明反应过程中不太可能生成多氟芳基自由基阴离子。
总结
德国马克斯-普朗克研究所Tobias Ritter教授课题组首次报道了通过光氧化还原脱羧策略,成功实现脂肪族羧酸的多氟芳基化反应。同时,该方法具有广泛的底物范围和良好的官能团耐受性。此外,通过对天然产物和药物的分子的后期修饰,进一步证明了该反应的实用性。



















No comments yet.