
本文作者:杉杉
导读
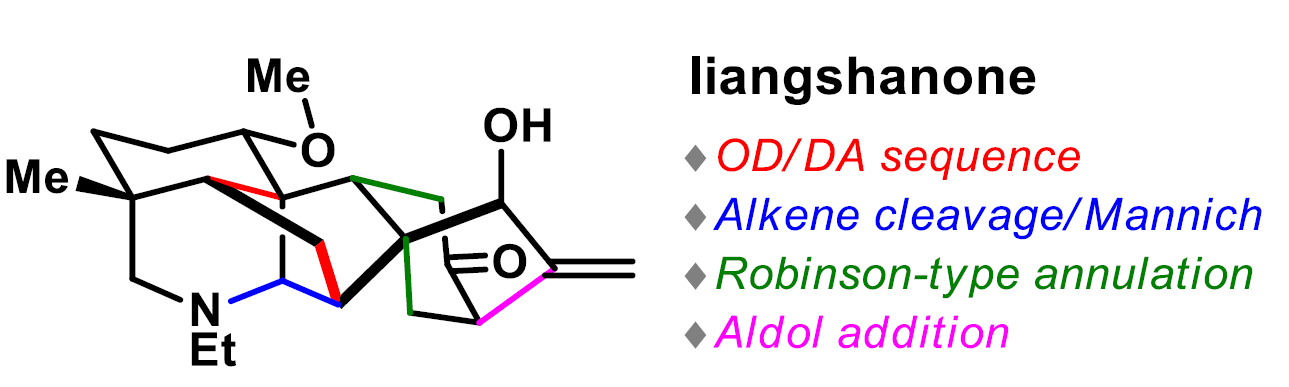
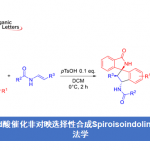
近日,四川大学华西药学院秦勇和刘小宇课题组在Angew. Chem. Int. Ed.上发表论文,首次报道了六环贝壳杉烷类生物碱(ent-kauranediterpenoid alkaloid)Liangshanone的全合成。通过几次关键的转换从而形成复杂的笼状骨架,涉及氧化去芳化/Diels-Alder(OD/DA)环加成、串联烯烃裂解/Mannich环化反应、Robinson型环化以及分子内的aldol反应。
Total Synthesis of Liangshanone
Hong-Xiu Huang, Fen Mi, Chunxin Li, Huan He, Feng-Peng Wang, Xiao-Yu Liu,* and Yong Qin*
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202011923 https://doi/10.1002/ange.202011923
正文
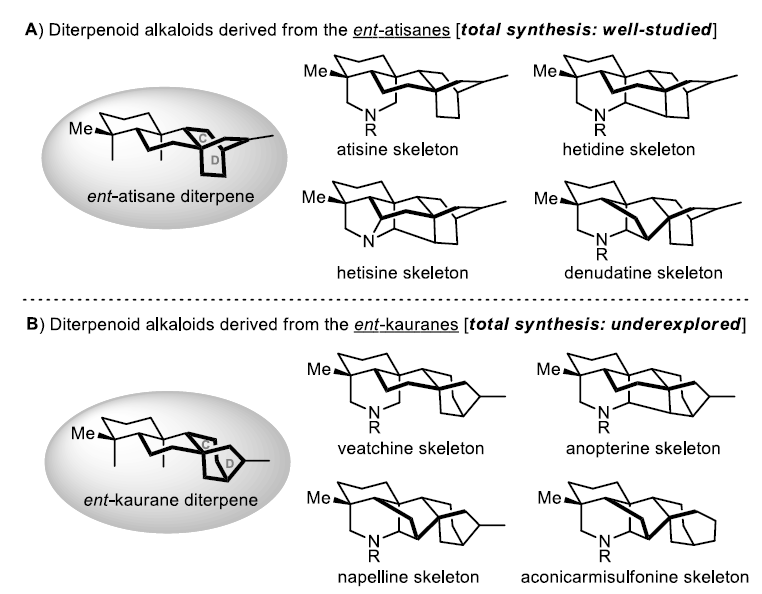
C20-二萜生物碱是一类十分具有吸引力的天然产物,由两类二萜前体衍生而来,即ent-atisanes和ent-kauranes。源自ent-atisane二萜(如atisines,hetidines,hetisines和denudatines;Figure 1A)的伪生物碱(pseudo-alkaloids)主要具有典型的双环[2.2.2]辛烷C/D环。相比之下,源自ent-kaurane二萜(如veatchines, anopterines, napellines, aconicarmisulfonines; Figure 1B)包含双环[3.2.1]辛烷C/D环。这些不同类型的C20-二萜生物碱均具有高度复杂的笼状结构,同时此类结构具有独特的生物活性,因此,整个ent-atisanes二萜类生物碱的全合成一直是研究的重点,虽然已实现多种创新的合成方法,但成功获得贝壳杉烷类生物碱(ent-kaurenoid alkaloids)仍然有限。
在各种类型贝壳杉烷类生物碱中,萘环(如1-3,Figure 1C)在结构上具有紧密的六环骨架,该骨架带有氮杂双环[3.3.1]壬烷(A/E环)、双环[2.2.1]庚烷(B/F环)和双环[3.2.1]辛烷(C/D环)。这些分子显示出一系列药理作用,涉及抗心律不齐、抗炎、抗焦虑等活性。迄今为止,Wiesner课题组报道了唯一以45步以上路线全合成纳哌啉型生物碱(napelline-type alkaloid)的例子。作为合成结构复杂的二萜生物碱的不懈追求,四川大学华西药学院秦勇和刘小宇课题组在此报道了Liangshanone的首次全合成(2)。
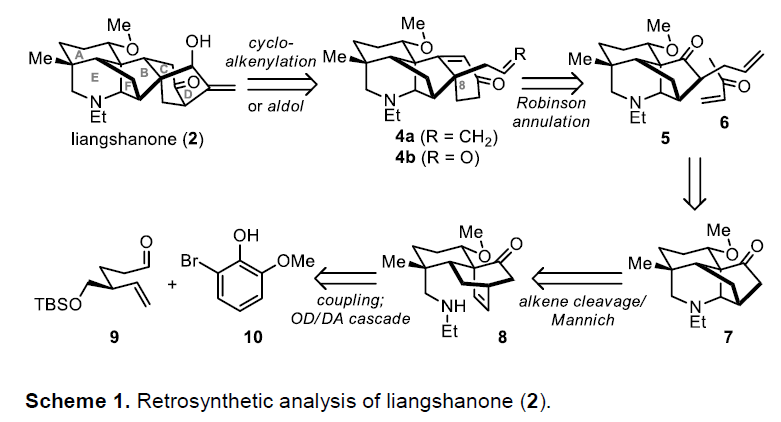
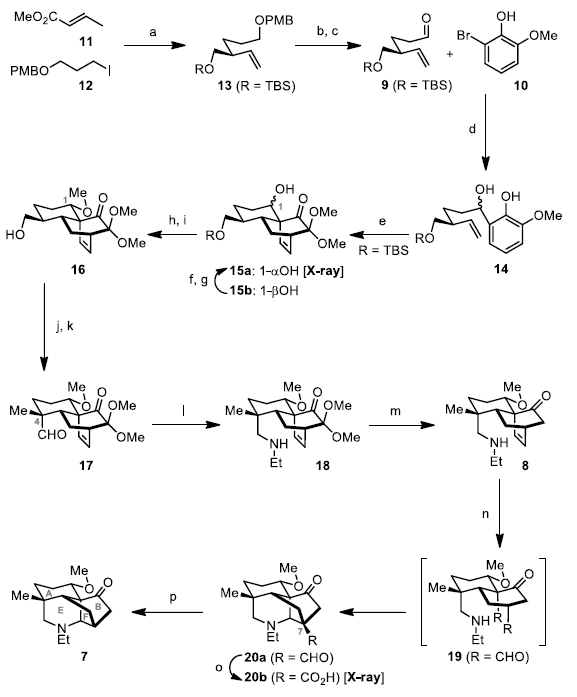
首先,作者对Liangshanone(2)进行了逆合成分析(Scheme 1)。若产物2中双环[3.2.1]辛烷部分(C/D环)断裂,则变为五环4a(分子内环烯基化)或4b(aldol加成)。而4a/4b可通过酮5和甲基乙烯基酮(MVK,6)经Robinson环化获得,同时可通过从四环中间体5的空间受阻较小的凸面来确保4a/4b中C8立体中心。反过来,5可以通过酮7的α-烷基化而产生。通过三环8的环烯烃氧化裂解和Mannich环化串联反应,获得四环7。而通过两个片段的偶联,然后通过氧化去芳化/Diels-Alder环加成(OD/DA)串联反应,可从醛9和溴酚10获得化合物8。
A/B/E/F四环中间体7的制备(Scheme 2)。在LDA/HMPA存在下,用已知的碘化物12与巴豆酸甲酯(11)进行去共轭烷基化,然后用LiAlH4还原羧酸酯并用TBSCl保护所得的伯羟基,即可一锅法获得60%收率的烯烃13。使用DDQ可将13中的PMB基团脱除,然后将其通过DMP氧化从而获得醛9。在-78 ºC下使用 nBuLi可使10与醛9反应获得二醇14(82%收率的非对映异构体,d.r. = 5:4)。接下来,在去芳化后,用PhI(OAc)2/MeOH处理苯酚14可生成二聚化的环加合物混合物,将其在均三甲苯中经180 ℃加热后,进行了串联Diels-Alder/分子内Diels-Alder环加成反应,从而获得可分离的非对映异构体15a(48%收率)和15b(39%收率)。同时,通过X射线晶体结构分析,进一步确定在C1上具有所需的α-取向羟基异构体15a,并且不需要的15b也可通过氧化和选择性还原转化为15a。随后,使用TBAF经脱甲硅烷基化反应,获得95%收率的醇16。接下来,16经DMP氧化,然后对所得醛进行非对映选择性α-甲基化,确保了C4季碳立体中心,从而以两步72%收率获得化合物17。用EtNH2•HCl/NaBH3CN进行醛17的还原胺化,得到胺18(88%收率)。随后,在THF/MeOH中用SmI2除去18中的两个甲氧基,得到8。
然后,将8在标准臭氧分解条件下(O3,-78 ℃;然后PPh3,0 ℃)反应,生成活性二醛中间体19,易在tBuNH2存在下进行Mannich反应,获得所需的产物20a(80%收率)。值得注意的是,该一锅法中,实现了一个碳碳键的断裂以及新的碳碳和碳氮键的形成,从而有效地将6/6/6三环8转化为6/5/5/6四环20a。此外,为了获得所需的产物7,需要将20a中C7处的多余碳除去。然而,各种金属介导的去甲酰化条件均未反应。因此,作者首先在标准的Pinnick氧化条件下将醛20a转化为酸20b(产率为95%),然后将酸20b与N-羟基邻苯二甲酰亚胺(NHPI)偶联,得到相应的酯,并在50 ℃下进行了镍催化的脱羧化反应,从而获得53%的产率的7。

要完成Liangshanone的全合成,剩下则为双环[3.2.1]辛烷C/D环的构建(Scheme 3)。首先,在酮7中引入了α-甲氧基羰基,以增强C8对随后的Michael加成的反应性。因此,使用LDA和Mander试剂对7进行处理,可获得β-酮酸酯21,在Cs2CO3条件下,将其与MVK反应,获得单一非对映异构体的二酮22(收率92%)。然后,与LDA进行反应,得到五环化合物23,若经脱水(Et3N,MsCl)过程则生成烯酮24。若通过还原(LiAlH4)和氧化(AZADO,DMAP,bpy,CuCl,air)这两个步骤,则以23%的总收率将获得醛25。用TMSOTf/Et3N原位保护25中的C9羟基和C12羰基,然后将反应混合物置于Wittig试剂Ph3P=CHOMe中,得到烯醇醚26。将上述粗产物用NaOH处理,进行脱硅基化和消除,得到不可分离的几何异构体27(Z/E = 5:1)。令人欣慰的是,可在40 ℃下使用TfOH可促进27中甲基烯醇醚的水解,同时进行分子内aldol加成,生成非对映异构混合物28(82%收率,d.r. = 1:1.3)。随后,通过三步官能化可将两种异构体均转化为酮29,包括催化氢化还原烯烃双键、缩酮化和仲羟基的氧化。而通过Petasis烯烃化,可进一步引入环外烯烃,实现29到30的转化(收率76%)。由于在用SeO2和叔丁基过氧化氢(TBHP)进行烯丙基氧化30的过程中观察到部分脱缩酮作用,因此在完全消耗30的条件下,将p-TsOH直接加入到反应混合物中,可除去缩酮保护基并生成31,即C15-Liangshanone。最后,用DMP和TFA氧化烯丙醇,然后在-78 ºC下使用NaBH(OMe)3对生成的酮进行区域和非对映选择性还原,从而获得天然产物Liangshanone(2)。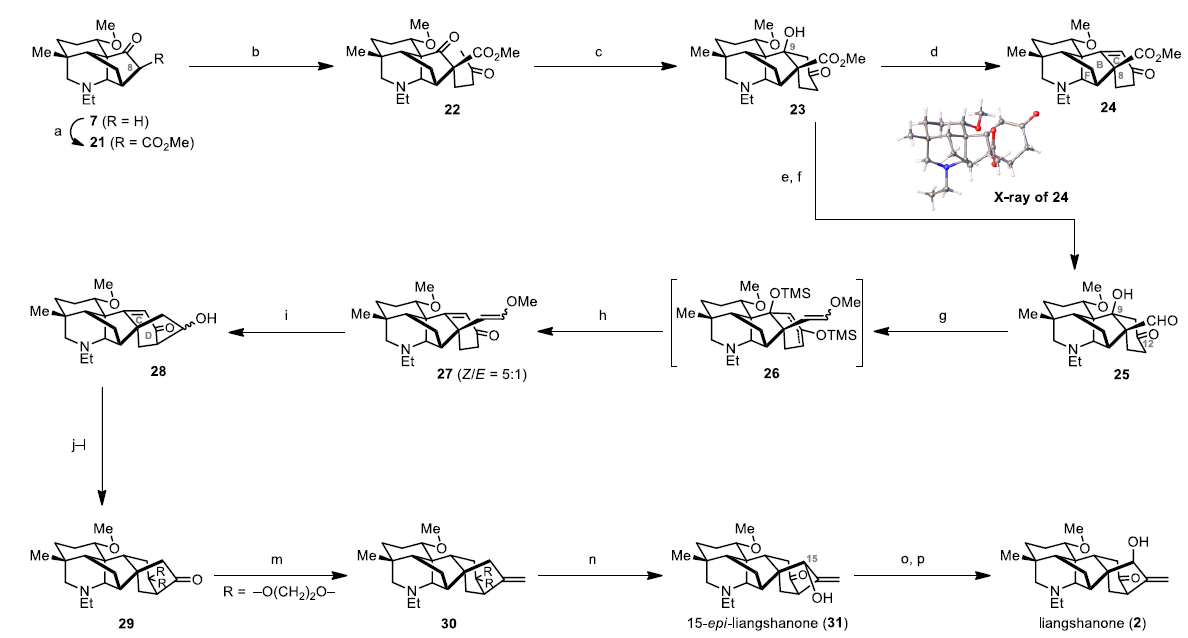

最后,作者对进行了三环中间体15a进行了对映选择性合成(Scheme 4)。使用Jørgensen-Hayashi催化剂33,进行醛32与甲醛的不对称α-羟甲基化反应,生成了环丙二醇34。将粗制的内酯中间体直接置于Wittig甲基化条件下,形成具有92% ee的高烯丙醇35(两步收率57%)。硅基化后(35至13),可将13转化为三环中间体15a。同时,15a可从己烷/二氯甲烷中重结晶,得到对映体富集的15a。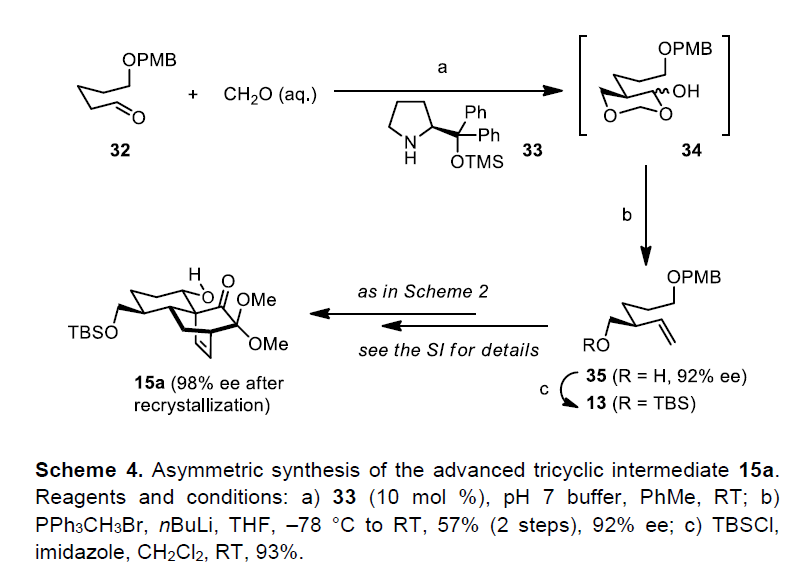
总结
四川大学华西药学院秦勇和刘小宇课题组首次报道了六环贝壳杉烷类生物碱Liangshanone的全合成。该反应涉及氧化去芳化/Diels-Alder(OD/DA)环加成、串联烯烃裂解/Mannich环化反应、Robinson环化以及分子内的aldol加成等关键反应步骤。此外,进一步证明了OD/DA策略在二萜生物碱的全合成中的实用性。















No comments yet.