
投稿作者 芃洋雪
1800s就发明的阿司匹林到1970s始终保守着一个巨大的秘密,使药物化学家躁动不安。阿司匹林(乙酰水杨酸)与环氧合酶COX发生共价键结合,是个不可逆的抑制剂,它将COX上的丝氨酸残基乙酰化,使环氧合酶失去了催化功能,从而阻止了后续的级联反应。这种不可逆的作用,引起了药物研发者的一丝担忧,它的强劲竞争者扑热息痛(对乙酰氨基酚)代谢产物也能共价结合到肝脏蛋白上引起毒性。在药物发现中避免药物永久结合到靶标上曾是金科玉律,然而时至今日这条铁律已被打破,共价小分子药物正在崛起。
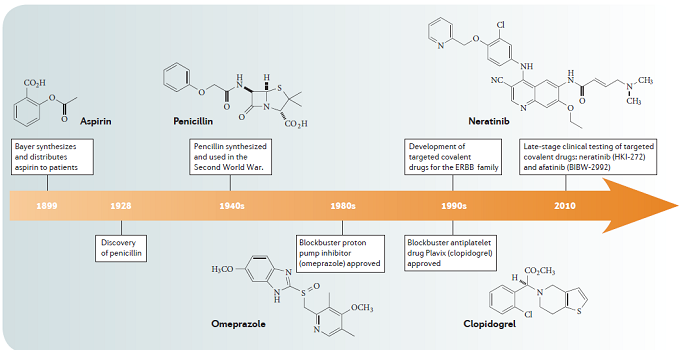
图1 一百多年来,已经有多个共价化合物上市。 图片来源:Nature Rev. Drug Discov.
2011年,Juswinder Singh等曾在Nature Rev. Drug Discov.上发文预测26个共价药物的年全球销售额将超过330亿美元,其中不乏氯吡格雷、兰索拉唑、噢美啦唑等重磅产品。
药物研发人员意识到共价结合,不仅能被允许,有时还是必需的手段。药物化学家已经不将希望寄托在偶然发现这种小概率事件上,转而合理设计共价化合物,过去5年的努力没有白费,现在正收获红利,5个有意设计的共价结合抗肿瘤药物已经上市。
根据锁钥理论,酶是锁,现在共价抑制剂作为钥匙效果更佳。Principia Biopharma的首席科学官David Goldstein认为这些分子能够一击必中,阻断酶的作用。等细胞再分泌出来这些酶通常需要数小时至数天,因此共价抑制剂的持续效果较长,患者可以服用更少的药物。别忘了事情都有两面性,酶的生命周期很长,而共价结合药物与酶的天然配体相竞争,因此药物设计时,还需注意应该使过多的药物尽快从体内排出。
非共价药物依靠的结合位点与其他酶家族成员的类似,而共价药物的结合点是独有的,因此共价抑制剂药物的选择性更高。这样患者接受药物量可以减少,副作用也随之减少,但这种作用也会引起抗性。理论上,相对于传统的非共价药物,这种理念设计出的小分子药物能够达到更高的选择性和更长的作用时间,提高药物的有效性和耐受性。适当的时候,一线药物科学家使用这种策略聚焦激酶抑制剂作为肿瘤药物。

Imatinib的结构。 图片来源:Chemistry World
激酶是很多生理活动的关键酶,人体中有超过500种激酶具有相似的活性位点,一度认为是不成药性,如果一种药物阻碍了肿瘤细胞的代谢,它也有可能阻碍其他的生理活动,从而导致严重的副作用。然而2001年酪氨酸激酶抑制剂Imatinib (Glivec),被批准治疗慢性粒细胞白血病以来,激酶抑制剂是重要的抗肿瘤药物。相比生命起来,肿瘤患者已经能够接受包括严重腹泻等不良反应。
选择性的针对少量的激酶仍然是药物化学家的主要挑战,Goldstein认为口服后药物浓度在胃部和上消化道很高,选择性差的激酶抑制剂会抑制消化道分泌的激酶,而导致严重副作用,甚至毒性。相比之下,酪氨酸激酶抑制剂通过共价结合半胱氨酸,达到了不可思议的选择性和高效性,应用这个策略靶向于成纤维细胞生长因子受体(fibroblast growth factor receptor)的PRN1371目前处于1期临床中。现在肿瘤患者已对非共价结合药物产生抗性。
合理设计共价抑制剂是成功的关键,这就需要相当精密的设计,对分子进行绘制,才能精确作用在一种酶的活性位点上,而不干涉其他酶的活性。需要充分理解共价抑制剂的亲电活性和生物学上依赖的亲核性指导设计。辉瑞的科学家分析共价抑制剂中的亲电性和谷胱甘肽、N-乙酰-L-赖氨酸的反应,来模拟半胱氨酸和赖氨酸的靶向反应。
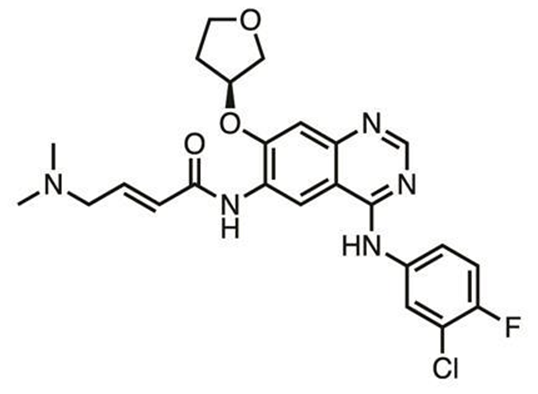
Afatinib的结构。 图片来源:Chemistry World
因为抑制剂永久性共价结合到蛋白质上,可以用质谱法研究详细情况,例如共价抑制剂TKIs和肿瘤细胞一起培养,将细胞粉碎后分析其中的蛋白质碎片,鉴定脱靶蛋白。Scripps的科学家还在研究共价抑制剂能否可逆。Kate Carrolls发现一些肿瘤细胞对勃林格殷格翰的Afatinib (Giotrif)产生抗性,靶向的EGFR中的半胱氨酸被氧化。肿瘤细胞中含有大量的活性氧,可以将半胱氨酸氧化为次磺酸形式的Cys-SOH,使其丧失亲核性,转化为亲电性。为此, Scripps的Florida开发共价抑制剂化合物库能和Cys-SOH反应。对科学家而言,最重要的是理解怎样将化合物分类,去粗取精,鸟枪换导弹。
尽管有这么大优势,共价药物目前仍然很少开发癌症之外的药物,除了几个偶然发现的老药。除了生死攸关的药物,因不可逆修饰脱靶蛋白带来的毒副作用被夸大了。高活性但选择性很差的共价抑制剂,会增加蛋白质分子量超过开启免疫效应而引发毒性问题。辉瑞的科学家们将放射性标记的共价药物和人类肝脏细胞,共同培养,评价可承受的共价结合负担。从中发现,使用多年的安全共价药物为小于10mg/天,以供优化共价药物朝向更安全的剂量。现在有多个公司将高选择性低剂量的药物推向临床。这些化合物表现出良好的安全性,虽然没有人能够计算或预测这些试剂的特异质毒性,但目前为止这些设计良好的新一代共价抑制剂表现出的毒性与可逆抑制剂类似。
与癌症的斗争还可以接受,但这意味着共价抑制剂在慢性病仍是挑战。University of California San Francisco (UCSF)的科学家正开发一种类似氰基丙烯酸酰胺的亲电化合物,它能与半胱氨酸成共价键,不同的是,它是可逆的(图2)。这样脱靶蛋白与之键合是暂时的,不同与永久性的改变,药物脱离后靶蛋白的结构和功能将恢复,因此可逆的共价结合不再与非共价结合药物存在显著差异。
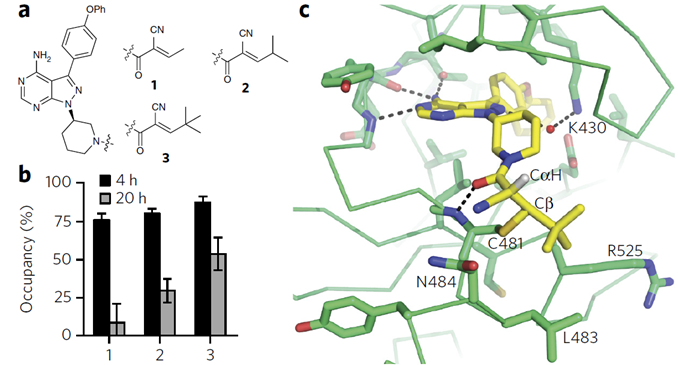
图2. a可逆的共价抑制剂结构, c共晶结构。图片来源: Nature Chemical Biology
例如布鲁顿酪氨酸激酶,BTK在免疫信号通路中起到重要作用,它引起类风湿性关节炎等疾病。正在进行2期临床的 PRN1008 作为可逆共价抑制剂治疗寻常性天疱疮,在口服后立刻抑制100%的BTK,然后它慢慢从靶蛋白中解离出来,在第2次服用之前下降至约有60-80%的抑制率。对Eli Lilly的药物化学家Bauer认为,现在共价抑制剂不仅仅局限于可逆反应的亲电试剂,还有其他鲜为人知的亲电体。Lilly也有一款治疗类风湿性关节炎的共价BTK抑制剂药在2期临床。
在共价抑制剂中,要注意化合物的外形和空间结构选择性,避免脱靶现象。即使是中等强度的亲电试剂,如丙烯酰胺也会发生烷基化反应。同其他药物一样,共价抑制剂也面临不被监管机构批准的高拒绝率。然而,这个新理念具有更坚实和深远的影响力。现在共价抑制剂的作用在药物发现领域非常重要,例如K-Ras(G12C)是一个 GTPase蛋白家族的突变蛋白,存在于约20%的肺癌患者中,之前曾认为无法成药,2013年 UCSF的Kevan Shokat发现共价化合物可以与突变的半胱氨酸反应(图3)。
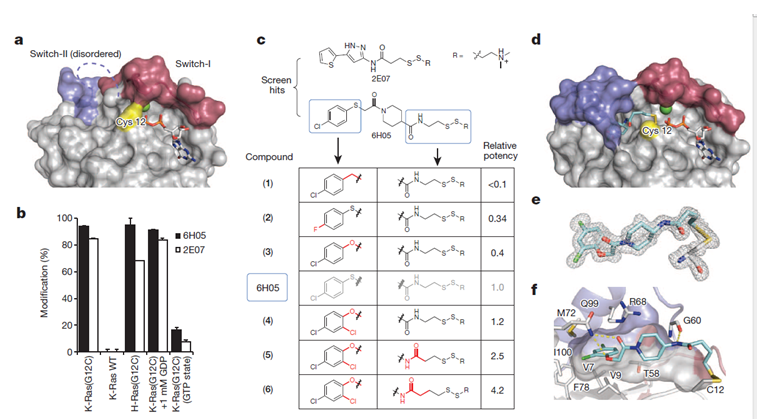
图3. a K-Ras(G12C)GDP的晶体结构,其中突变的Cys12为黄色;c 6H05的类似物及构效关系;d化合物6和K-Ras(G12C)GDP的共晶结构。图片来源:Nature
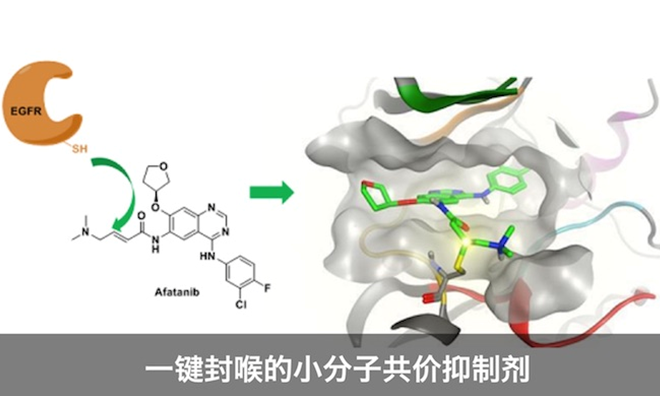
现在共价抑制剂近年来在很多领域取得了成功,共价化学开发的激酶抑制剂,可以和传统的可逆抑制剂一争高下,它一键封喉的能力使其在化学药物中占据一席之位。
(文章编译Chemistry World的评论:Drugs, the permanent way)
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!


















No comments yet.