
本文作者:杉杉
导读
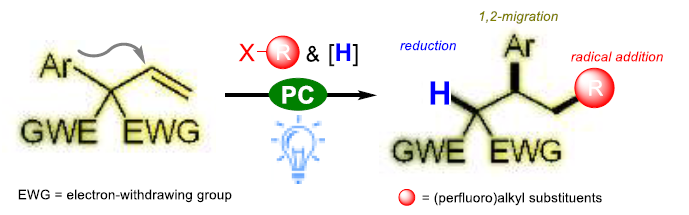
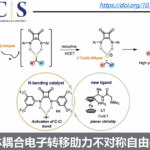
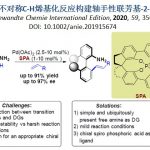
近日,南京大学史壮志教授课题组在Angew. Chem. Int. Ed.上发表论文,报道了一种有效的光催化方法,可实现邻季碳烯烃的全氟烷基化(perfluoroalkylation)反应,涉及1,2-芳基迁移。同时,可通过一步直接合成具有价值的多取代全氟烷基化化合物(其他方法的制备则具有挑战)。机理研究表明,通过光诱导产生的烷基自由基与烯烃加成,经1,2-芳基迁移,获得具有两个吸电子基团的碳自由基,其再与氢供体进一步还原以完成反应。
Radical Addition Enables 1,2-Aryl Migration from Vinyl-Substituted All-Carbon Quaternary Center
Zexian Li, Minyan Wang, and Zhuangzhi Shi
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202010839 https://doi.org/10.1002/ange.202010839
正文
重排反应(操作简便、高原子经济性)是化学合成领域中最具有吸引力的转化方法之一,可用于构建多种新型骨架的有机化合物。在此类反应中,自由基介导的官能团迁移已受到越来越多的关注,而其中1,2-重排是该领域的热点。Urry和Kharasch等早在1944年发现了以β-芳基碳为中心的第一个自由基1,2-迁移(neophyl重排),然而,由于底物范围窄,反应缓慢以及苛刻的反应条件,使得该反应难以应用于有机合成中。
在过去的十年中,许多课题组已开发了一系列烯烃底物,用于自由基诱导的1,2-重排。如利用已报道烯丙醇的半频哪醇型重排(semipinacol-type rearrangement)来制备β-官能化的酮衍生物(Figure 1a)[2];通过使用Et3B/空气[3],可见光[4]或镍催化剂[5]实现乙烯基硼酸酯配合物的1,2-金属盐的重排(Figure 1b)。受这些结果的启发,该课题组还开发了光氧化还原烯烃1,2-硼基迁移,通过自由基加成生成偕-双(硼基)烷烃[6]。此外,Studer课题组最近报道了一种使用烯丙基硼酸酯的1,2-硼基迁移基团,用于合成1,3-二官能团化产物(Figure 1c)[7]。在此,南京大学史壮志教授课题组描述了一类带有邻季碳烯烃底物,涉及自由基加成、1,2-芳基迁移、还原的过程(Figure 1d)。
在有机化合物中引入全氟烷基取代基通常会导致其性质(如极性,溶解度,构象行为和代谢稳定性)发生很大变化。烯烃的全氟烷基化一直是将全氟烷基取代引入有机分子的最有效方法之一。与以前文献报道的不同,该反应通过全氟烷基-自由基加成至烯烃双键上,再经1,2-芳基迁移后再被氢供体进一步还原。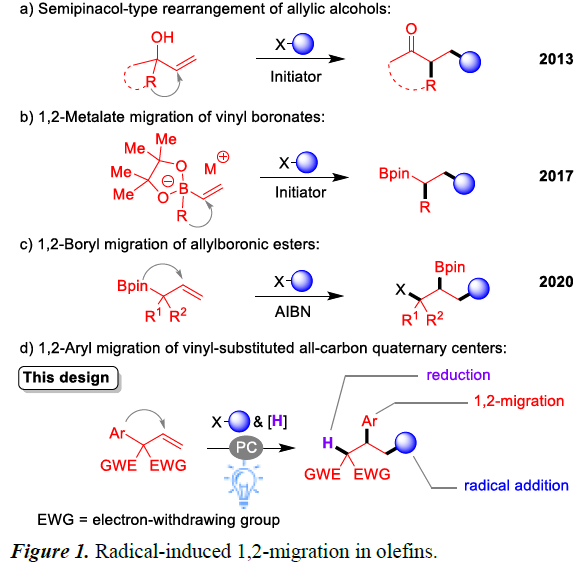
作者以烯烃1a与碘化物I-nC4F9(2c)作为模型反应底物,进行了相关反应条件的筛选(Table 1)。反应的最佳条件为:使用 1.0 mol%fac-[Ir(ppy)3]作为光催化剂,2当量DBU作为碱,在TFE溶液中(包含2.0当量的TMDEA)于室温蓝色LED照射12 h后,可获得78%收率的3aa(entry 1)。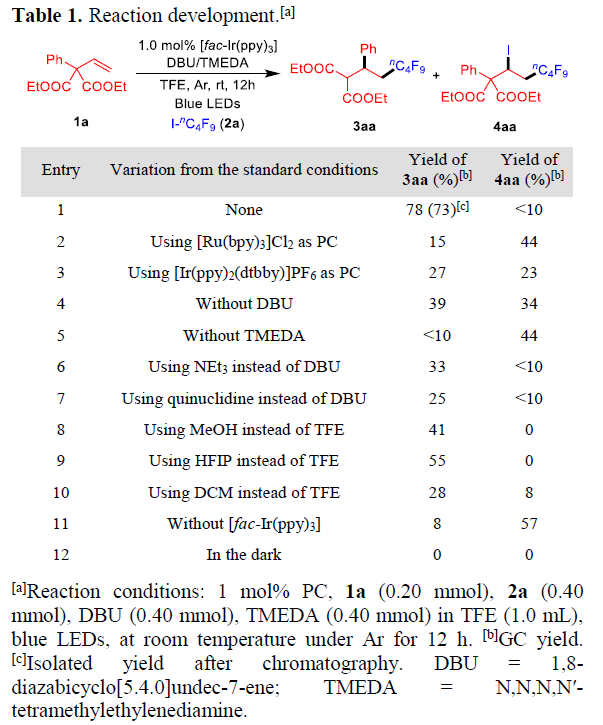
在获得上述最佳反应条件后,作者首先对烷基卤底物2进行了扩展(Table 2)。具有长链的烷基碘(如2b、2c)可与体系兼容。同样,二氟甲基化的烷基溴化物2d-2e也是合适的底物,但单氟甲基化的底物2f仅获得较差的非对映选择性产物3af。而具有更长全氟链烷基碘化物2g-2i,具有三级脂肪族全氟烷基碘化物2j-2k,以及其它烷基卤化物,如2-碘乙酸乙酯(2l)和2-碘乙腈(2m),均可获得所需的目标产物3ag-3am。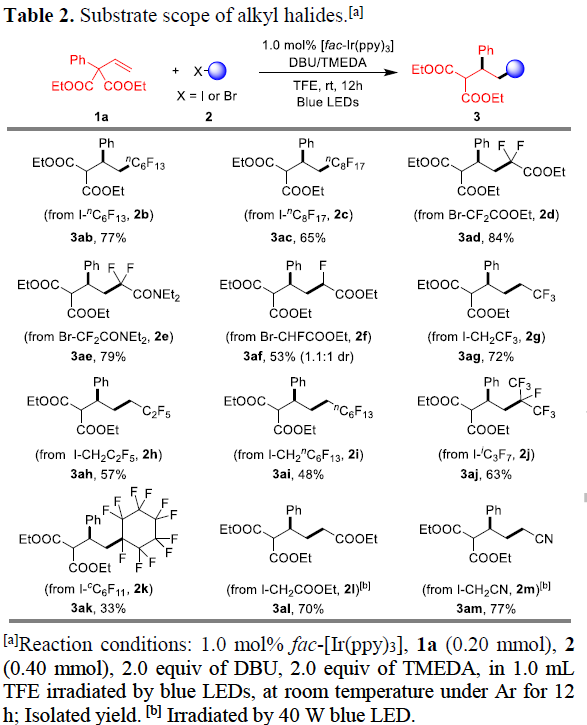
同时,作者也将不同氟代试剂分别引入体系中(Scheme 1)。如使用廉价且丰富的CF3I(2n)与烯烃1a在标准条件下反应,获得69%收率的产物3an。令人欣慰的是,当使用TMEDA•2CF3I(2n’)时,收率可提高至75%。此外,使用二氟试剂2o(Hu′s试剂)[8a]与烯烃1a进行二氟甲基化时,获得56%收率的产物3ao。然而,单氟试剂2p[8b]反应性较低,可能是由于缺乏自由基稳定基团所致。
紧接着,作者对烯烃底物1进行了扩展(Table 3)。芳基对位带有供电子和缺电子的取代基时(如OMe、Ph、F、Br、CN),均可获得相应的产物3ba-3fa(54-89%)。芳基间位带有CF3、COOEt和Cl取代基时,也可获得相应的产物3ga-3ia(69-86%)。具有萘基、杂芳基取代时,也可获得产物3ja-3ka(67-80%)。令人高兴的是,这种允许烯烃重排的策略包含多种EWGs,如1l和1m,均可实现相应的转化。然而,具有大体积的巴比妥酸底物1n与2a反应时,获得主要的原子转移自由基加成产物4na以及痕量的目标产物3na(<10%)。值得注意的是,使用Hu’s试剂2o与烯烃1n反应时,若底物中不存在碘原子,则重排转化率更高,获得51%收率3no。此外,仅含一个EWGs的1o,重排非常缓慢,其中原子转移自由基加成化合物4oa为主要产物。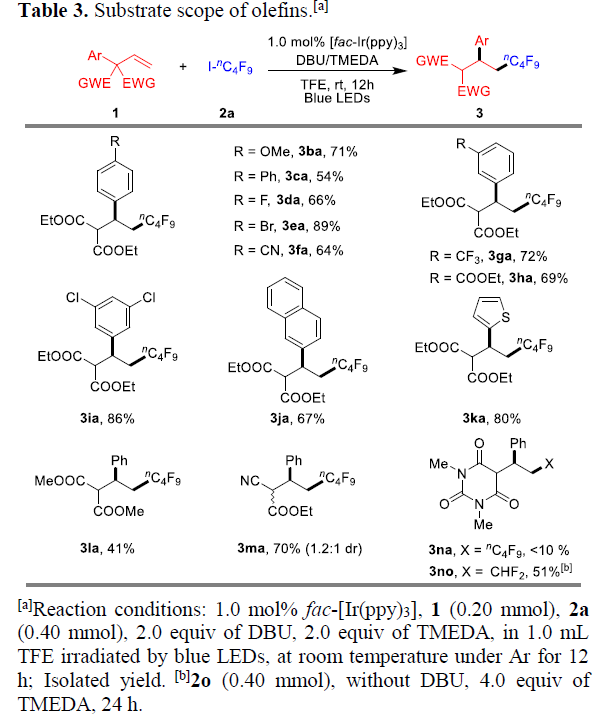
紧接着,作者对该反应实用性进行了研究(Scheme 2)。首先,三氟甲基化反应可以扩展至2.0 mmol规模,以58%的收率获得产物3an。该化合物可选择性地还原成二醇5(89%)或脱羧成酯6(81%)。同时,经Michael加成反应(7),亲核取代反应(8-9)和Aldol反应(10),可生成具有季碳中心的产物7-10(73-84%)。同时,在碱性条件下,使用NBS可将产物3an溴化,以中等收率获得化合物11(50%)。
为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 3)。首先,反应中形成的副产物4aa在标准条件下未形成目标产物3aa,从而排除了4aa作为中间体的可能性(Scheme 3a)。据Sodeoka所报道[9],化合物12-13与更长的烯基链反应形成环化产物14和15,而为观察到1,n-芳基迁移产物(Scheme 3b)。当使用d1-MeOH作为溶剂进行烯烃1a和2a的反应时,D标记几乎全部结合到3aa中。通过13C NMR进一步检测,反应混合物在90.4和54.8 ppm处产生两个信号峰,这与反应中d1-甲氧基甲醇(16)的形成相一致。而且,d1-MeOH与3aa反应不会形成氘代产物D-3aa(Scheme 3c)。这些结果表明,在该反应过程中发生了H/D交换过程,并且醇溶剂充当了产物氢原子的供体。此外,“明/暗”实验进一步证实可见光是反应的必要组成部分。最后,对烯烃1a与I-nC4F9(2a)反应的量子产率进行测量,得出Φ= 1.8,表明生产性的短寿命自由基链增长过程与主要的光氧化还原循环同时进行。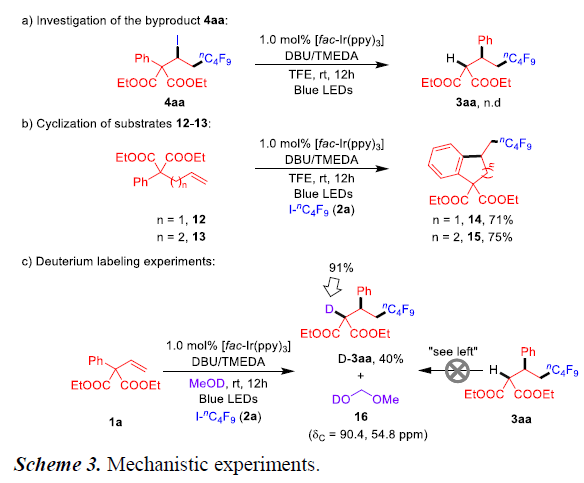
根据上述的实验,作者提出了一种可能的反应机理(Figure 2)。首先,可见光对IrIII配合物进行光激发,得到具有氧化还原活性的激发态*IrIII,它与烷基卤化物2进行单电子转移(SET)以产生IrIV配合物和烷基自由基I。IrIV配合物与叔胺进行SET反应以回收IrIII配合物并生成胺自由基阳离子A。紧接着,配合物A与TFE溶剂进行氢原子转移,从而释放出胺盐B和碳自由基C。进一步的β-H消除可形成CF3CHO和一个氢原子基团。通过对反应混合物中NMR分析,可能是有CF3CHO和TFE形成的三氟乙氧基半缩醛17。同样,使用d1-MeOH作为溶剂会生成氘代产物D-3aa和HCHO,根据实验结果,它们可以进一步与d1-MeOH反应,生成化合物20。此外,形成的自由基I与烯烃1加成,获得烷基自由基II,经螺[2.5]辛二烯基自由基过渡态III的1,2-芳基迁移后,形成自由基IV。最后,自由基IV与氢自由基结合,生成目标产物3。同时,在催化循环之外,烷基II可与卤代烷2反应,形成ATRA产物4。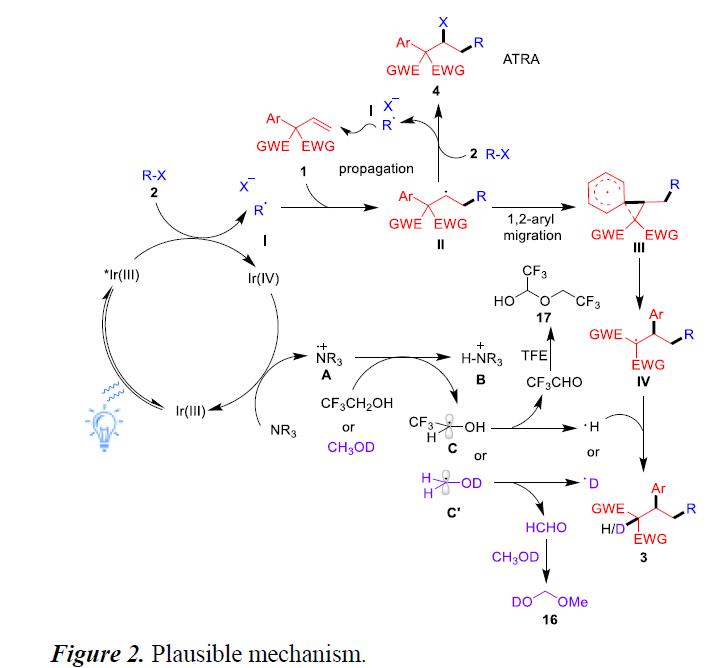
总结
南京大学史壮志教授课题组开发了一种可见光光氧化还原催化策略,以邻季碳烯烃为底物,可高效且发散的合成多种多取代全氟烷基化合物。涉及自由基加成、1,2-芳基迁移、还原等过程,以及一个C-C键断裂同时形成一个C-H和两个C-C键的过程。
参考文献
[1] W. H. Urry, M. S. Kharasch, J. Am. Chem. Soc. 1944, 66, 1438.[2] a) T. J. Snape, Chem. Soc. Rev. 2007, 36, 1823; b) B. Wang, Y.-Q.Tu,Acc. Chem. Res. 2011, 44, 1207; c) Z.-L.Song, C.-A.Fan, Y.-Q.Tu,Chem. Rev. 2011, 111, 7523.
[3] a) M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld, A. Studer,Science 2017, 355, 936; b) C. Gerleve, M. Kischkewitz, A. Studer,Angew. Chem. Int. Ed. 2018, 57, 2441; Angew.Chem. 2018, 130,2466.
[4] M. Silvi, C. Sandford, V. K. Aggarwal, J. Am. Chem. Soc. 2017, 139,5736.
[5] G. J. Lovinger, J. P. Morken, J. Am. Chem. Soc. 2017, 139, 17293.
[6] B. Zhao, Z. Li, Y. Wu, Y. Wang, J. Qian, Y. Yuan, Z. Shi, Angew.Chem. Int. Ed. 2019, 58, 9448; Angew.Chem. 2019, 131, 9548.
[7] K. Jana, A. Bhunia, A. Studer, Chem2020, 6, 512.
[8] a) Y. Zhao, W. Huang, L. Zhu, J. Hu, Org. Lett. 2010, 12, 1444; b) Q.Liu, X. Shen, C. Ni, J. Hu, Angew.Chem. Int. Ed. 2017, 56, 619;Angew.Chem. 2017, 129, 634.
[9] a) H. Egami, R. Shimizu, S. Kawamura, M. Sodeoka, Angew. Chem. Int. Ed. 2013, 52, 4000; Angew.Chem. 2013, 125, 4092; b) S.Kawamura, M. Sodeoka, Angew. Chem. Int. Ed. 2016, 55, 8740; Angew. Chem. 2016, 128, 8882.
















No comments yet.