
本文作者:杉杉
导读
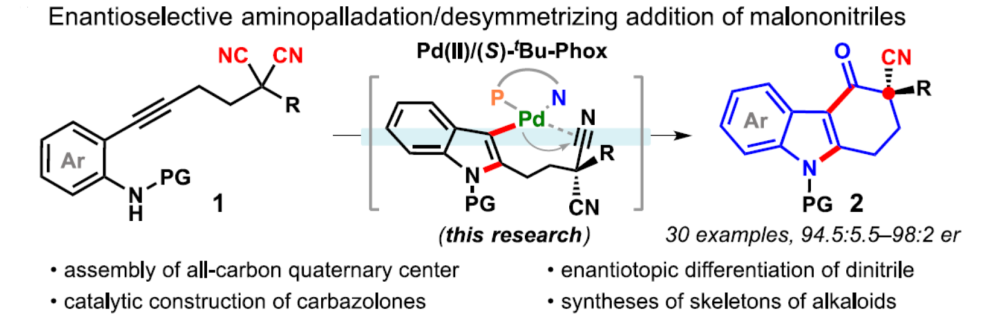
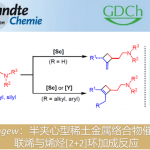
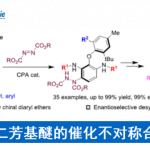
近日,武汉大学刘文博课题组在JACS上发表论文,通过使用含有炔烃的丙二腈底物(1),实现了Pd(II)催化氨基环化和去对称化(desymmetrizing)腈加成串联反应,从而合成具有α-季碳立体中心咔唑酮(carbazolones)衍生物(er高达98:2)。同时,该反应可通过一步操作构建两个新环和一个季碳中心。此外,通过产物后期的衍生化以及相关天然产物的合成,进一步证明了反应的实用性。
Enantioselective Synthesis of α‑All-Carbon Quaternary Center-Containing Carbazolones via Amino-palladation/Desymmetrizing Nitrile Addition Cascade
Xu-Dong Hu, Zi-Hao Chen, Jing Zhao, Rui-Ze Sun, Hui Zhang, Xiaotian Qi, and Wen-Bo Liu*
J.Am. Chem. Soc.ASAPDOI:10.1021/jacs.1c00840
正文
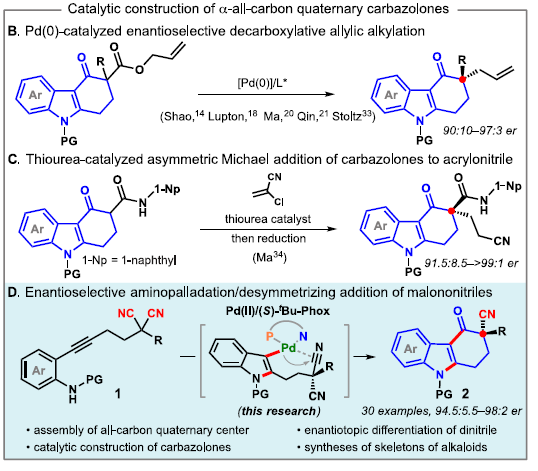
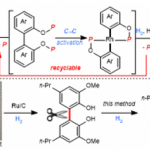
咔唑酮是生物学中一类重要的骨架。目前,利用咔唑酮作为关键中间体,已实现超过10种Aspidosperma和Kopsia植物碱的全合成(Scheme 1A)。这些研究具有统一特征,即充分利用C20的全碳立体中心,从而实现立体选择性的控制。然而,仅有两种类型的不对称催化方法被报道,即Pd(0)催化脱羧烯丙基烷基化反应和硫脲催化Michael加成反应(Scheme 1B,C)。2013年,Shao[1]和Lupton[2]等实现了对映发散性Pd(0)催化的脱羧烯丙基烷基化反应,从而构建具有季碳中心的咔唑酮(Scheme 1B)。随后,其他的课题组也报道了相似的反应合成具有不同环外取代基的咔唑酮,如Ma[3]、Qin[4]、Stoltz[5]、Song和Chang[6]。最近,Ma等[7]还实现了硫脲催化咔唑酮(含酰胺取代)和2-氯丙烯腈的不对称Michael加成/还原反应。虽然已取得一定的成果,但此类方法需预先进行立体化学合成咔唑酮(只能引入有限的官能团),从而导致方法受到限制。因此,开发一种新型串联反应以构建具有α-全碳四元中心的咔唑酮骨架具有重要的意义。此外,受炔烃钯化反应和碳-杂原子不饱和键插入串联反应的启发,作者设想了一种去对化的策略,通过易合成的炔基化丙二腈为底物,经反式-胺钯反应形成芳基钯(II)中间体,随后经水解后插入腈基,从而获得具有α-季碳中心的咔唑酮衍生物(Scheme 1D)。
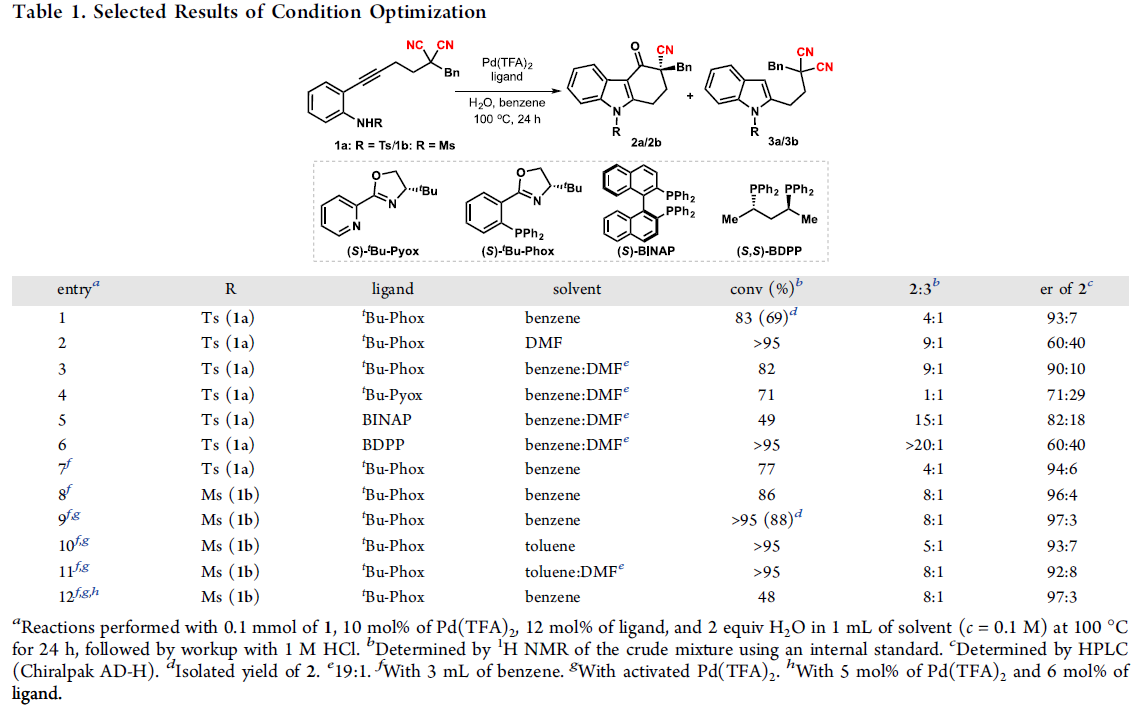
首先,作者以含炔烃的丙二腈1a和1b作为模型底物,进行了相关反应条件的筛选(Table 1)。当使用含有Ms基团的1b为底物,以Pd(TFA)2和(S)-tBu-Phox原位反应生成化合物为催化剂,可在甲苯溶剂中反应,获得咔唑酮2b,收率为88%,er为97:3。
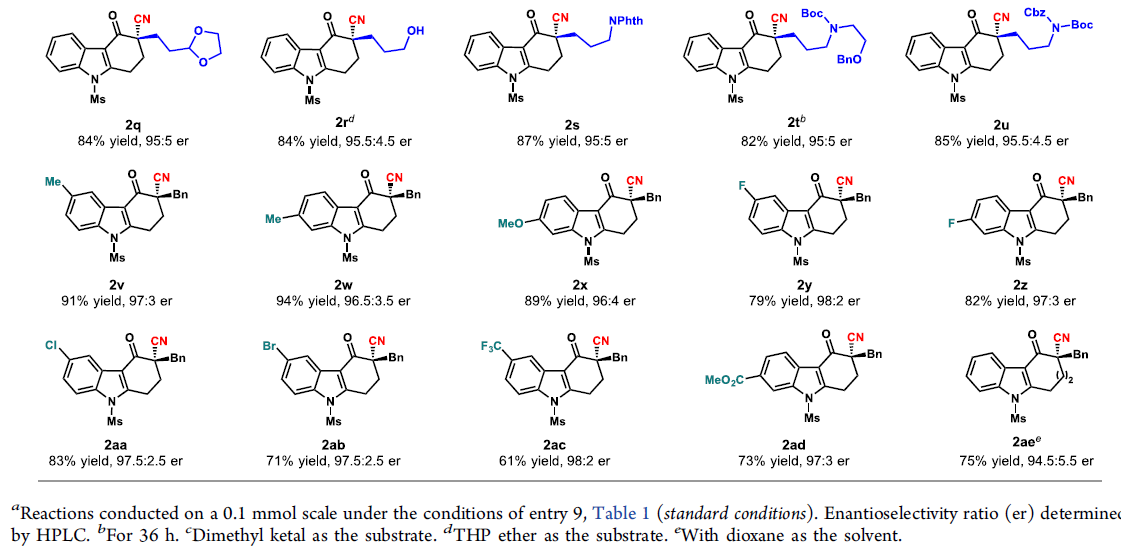
在获得上述最佳反应条件后,作者开始对底物1进行了扩展(Scheme 2)。首先,在对底物中α-位官能团耐受性研究表明,具有(杂)芳基甲基(2b–2g)和烯丙基取代基(2h)的丙二腈是可行的底物,获得80-94%收率的产物。而含炔烃的底物(2i)具有较低的反应性,收率为59%。一系列具有简单烷基取代基的底物,均可顺利反应,获得较高收率的产物2j–2n。值得注意的是,该反应也可获得72%收率的α-苯基咔唑酮2o。同时,α-位取代含有一些活性基团时,如酮、乙缩醛、醇、酯等,均与体系兼容,获得产物2p–2u。其次,在对底物中芳基环的耐受性研究表明,具有给电子取代基(Me-和MeO-)的底物在标准条件下均能顺利反应,获得89-94%收率的咔唑酮2v–2x。卤化物(F-、Cl-、Br-)、三氟甲基和酯取代的底物(2y–2ad)也与体系兼容,但收率略低。最后,该反应也可合成吲哚稠合的七元环产物2ae,收率为75%。然而,其它一些具有较短或较长碳链等的底物无法获得相应的产物。值得注意的是,上述产物均具有较高的对映选择性。
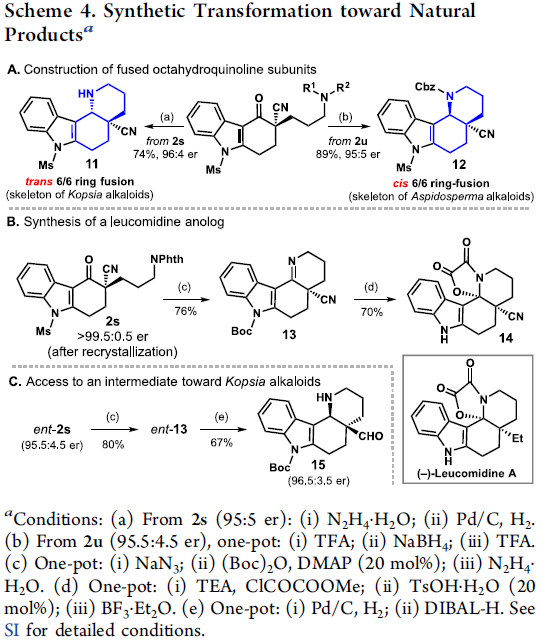
紧接着,作者对反应的实用性进行了研究(Scheme 3)。在标准条件下,2b的克级实验,收率为87%,er为96:4。同时,相应产物2可进行多种后期衍生化。2b经NaN3处理可去除Ms基团,获得当量收率的无保护咔唑酮4。在Virgil和Grubbs[8]报道的铂催化水合反应之后,具有位阻的腈基很容易转化为酰胺5,收率95%。2b经水合和Hofmann重排,可获得83%收率的异氰酸酯6。2p经分子内Aldol缩合,可获得88%收率的环化产物7。2r经PPh3/CBr4、NaN3和TiCl4处理后,可获得67%收率的螺-四唑8。2b通过一锅法NaBH4和Et3SiH还原后,可获得91%收率的β-季四氢咔唑9。最后,还原2c中的羰基,然后进行酸促进Friedel-Crafts反应,以极好的收率获得环稠合化合物10。值得注意的是,在所有衍生化反应中,er值均不受影响。

此外,含有α-季碳中心咔唑酮作为一些天然产物的关键中间体,可通过咔唑酮2s和2u高效的合成单萜吲哚生物碱核心的顺式和反式稠合的八氢喹啉骨架(Scheme 4A)。如通过水合肼除去2s中的邻苯二甲酰基,可实现自发环化形成亚胺,再经Pd/C和H2氢化反应以形成反式稠合产物11(Kopsia生物碱核心骨架),收率为76%。2u通过一锅法的酮还原和酸促进环化,可获得顺式稠合产物12(即Aspidosperma生物碱的骨架),收率为89%。其次,从咔唑酮2s开始,也可直接合成了螺环14,一种Leucomidine A的类似物(Scheme 4B)。最后,ent–2s经脱保护/环化反应,获得80%收率的ent–13,再经亚胺和腈的还原,即可获得67%收率的四环稠合醛化合物15,可作为合成Kopsia生物碱、Kopsinidine C和Kopsinitarine E的关键性中间体(Scheme 4C)。
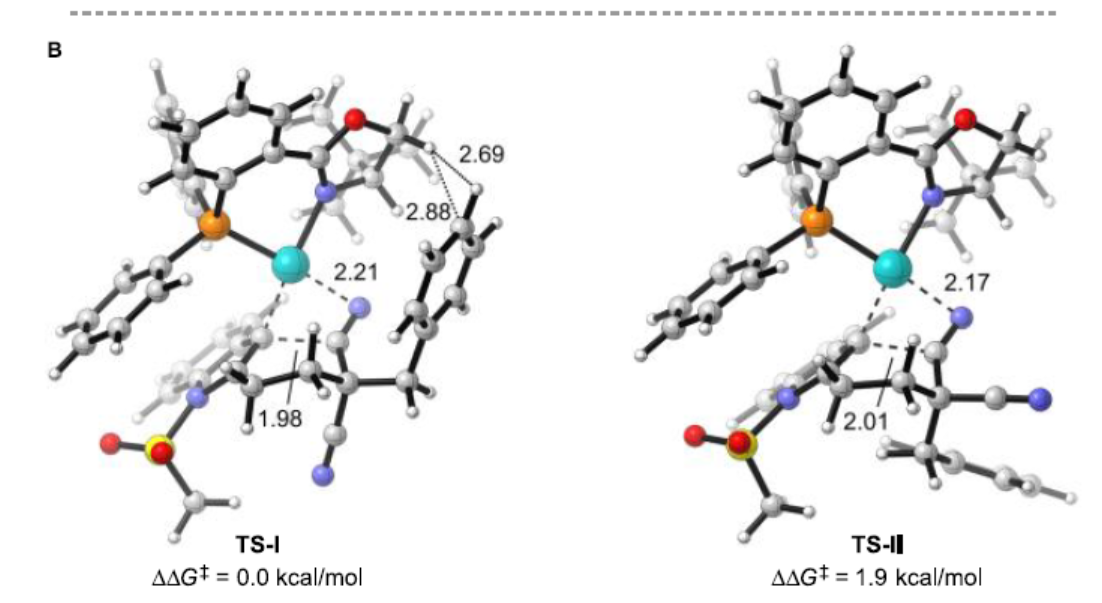
根据相关文献的查阅[9],作者提出了一种可能的反应机理(Scheme 5A)。首先,Pd(II)配合物I与1b中的炔烃配位,经反式-胺钯化反应形成吲哚基钯(II)配合物III。随后,配合物III中经分子内亲核加成生成配合物IV,经质子化,形成亚胺中间体V并再生催化剂I。其中,中间体V经进一步酸性水解,即可获得咔唑酮2b。此外,在条件优化过程中发现,配合物III经质子分解可形成副产物3b,也暗示了另一种可能的机理,即钯(II)催化3b经中间体VI的Friedel-Crafts反应形成IV,但将3b置于标准条件下不会生成2b,从而排除这种可能性。此外,作者还对腈基的插入进行了DFT计算,以阐明二腈基的对映异构分化(Scheme 5B)。计算结果表明,TS-I的能垒比TS-II低,这是由于体积较大的Bn基团位于半椅状环的空间较少的位置导致。
总结
武汉大学刘文博教授课题组开发了一种不对称钯(II)催化氨基环化和去对称化腈加成串联反应,可合成高收率和高对映选择性且含有α-全碳四级立体中心咔唑酮衍生物。值得注意的是,咔唑酮可作为一些生物碱的通用结构单元和核心骨架(如Leucomidine A)。此外,通过产物后期的衍生化,进一步证明了反应的实用性。















No comments yet.