
今年年初,小编介绍了一篇碱金属催化的C-H键的直接硅基化反应,不知道大家是否还记得(记事:什么?碱金属发生的硅基化反应!)。那篇文章打破了常规的使用过渡金属催化剂的传统,只是利用碱金属(KOt-Bu)就可以达到硅基化的目的,是一个十分有突破性的研究。
最近,日本理化研究所侯教授(我们chem-station做过侯老师的专访-筑梦化学键的自由切断、自由构建,有兴趣的可以点开链接回顾下)在这个基础上更进一步,直接不需要使用任何金属就可以达到C-H硅基化反应。
本文小编整理了一下最近几年的具有代表性的三种直接硅基化反应手法,并且重点介绍一下侯老师的这第三种方法:硼催化的C-H硅基化反应。
Ma, Y.; Wang, B.; Zhang, L.; Hou, Z. J. Am. Chem. Soc., 2016, 138, 3663. DOI:10.1021/jacs.6b01349
第一种方法:利用过渡金属催化剂催化的手法
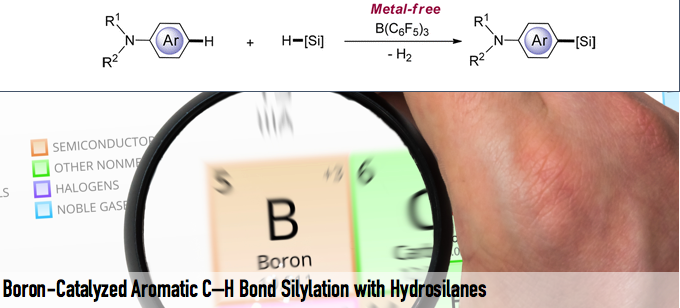
过渡金属催化剂催化的芳环的C-H硅基化反应,是近年来比较热门的研究[1]。在多种过渡金属催化的硅基化反应中,小编认为比较具有代表性的是2014年,美国加州伯克利分校的Hartwig教授等人利用Rh催化剂催化的C-H硅基化反应[2]。
如下图1(a)所示,该反应使用L1作为金属的配体、(TMSO)2MeSiH作为硅基化反应试剂,另外环己烯为氢气受体,实现了芳香环上位阻最小得位置的硅基化反应。在这之后,他们又开发出了利用新的Ir催化剂催化的C-H硅基化反应[3]。与Rh催化剂相比具有更广泛的官能团兼容性,能够适用于含有卤素的底物或者杂环底物(图 1(b))。
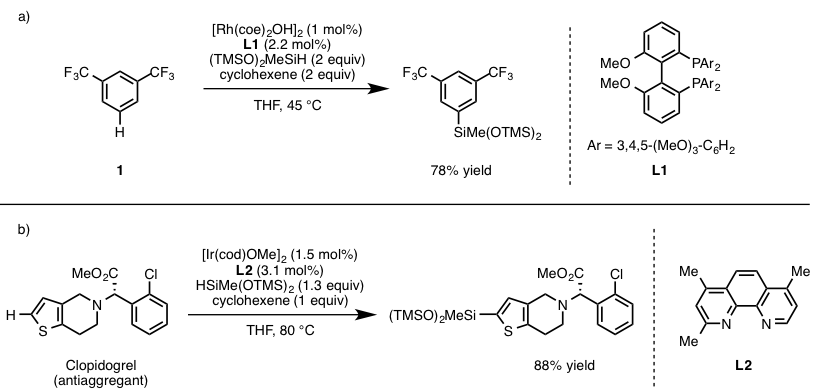
图1. 过渡金属催化剂催化的C-H硅基化反应
第二种手法:利用碱金属KOt-Bu的手法
2015年,美国加州理工的Stoltz和Grubbs教授的研究组报道了仅仅使用KOt-Bu实现的芳香杂环的C-H硅基化反应(记事:什么?碱金属发生的硅基化反应!)[4]。该方法仅仅利用催化量的KOt-Bu,利用Et3SiH作为硅基化试剂,对N-甲基吲哚实现了2位选择性的硅基化(图 2)。
本反应被认为是通过自由基反应机理,可以实现尤富电子的芳香杂环化合物的硅基化反应。
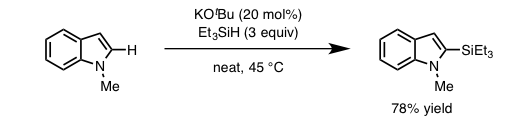
图2. KOt-Bu催化的芳香杂环化合物的C-H硅基化反应
第三种手法:利用硼催化剂的手法
在最近侯等人利用B(C6F5)3催化活化Si-H键,作为与上述第一第二种不同的新手法,实现了C-H硅基化反应[6]。
关于B(C6F5)3
含有一个空轨道的三取代硼化合物具有很高的路易斯酸性。在通常的硅基化反应中,B(C6F5)3与硅试剂可以形成特殊的复合体X(图3)[5]。也就是说,B(C6F5)3与硅试剂的Si-H之间存在H对B配位后形成了复合体X。这样就导致Si显正电性,到目前为止,通常也是利用这个特点,进行各种硅基化反应[5]。例如下图所示,复合体X可以作用于醇类化合物,就是由于这个因素,伴随着氢气的产生进行的醇类的硅基化保护[5(a)]。
图3 氢对硼配位形成复合体X
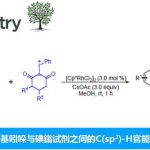
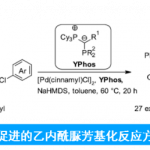
侯教授等人利用该常用的硼化合物,开发出了下图所示的新的硅基化反应。如下图所示,以N,N-二甲基苯胺为底物,利用催化量的B(C6F5)3,在二苯基硅烷的作用下,成功实现了底物的对位选择性硅基化(图4)。
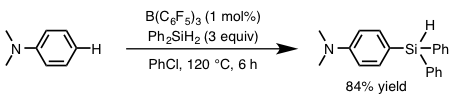
图4. 硼催化的芳香环的对位选择性硅基化
[假定的反应机理]
论文中假定的反应机理如下图所示(图5)。反应起始阶段,如上述所述,首先B(C6F5)3与硅基化试剂作用形成正电性的硅烷复合体A。然后这个活性中间体A与N,N-二甲基苯胺发生加成反应,然后经由中间体B的脱氢气作用,得到产物,并且使得B(C6F5)3得到再生,完成整个催化循环。
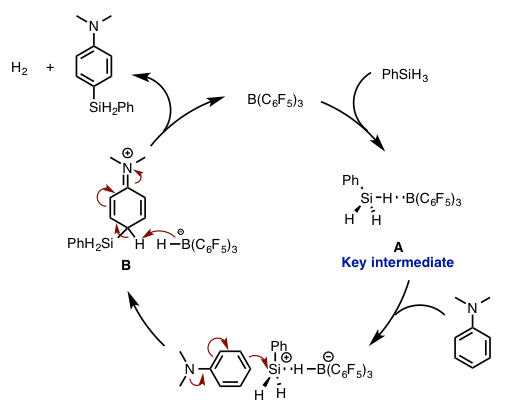
图5. 假定的反应机理
[本反应的特征]
本反应是N,N-二甲基苯胺的对位选择性硅基化反应,同样适用于各种苯胺衍生物。同时,对于硅基化试剂来说,也是很有广泛性。在利用氯代硅试剂的情况下,如下图所示,可以进一步转化成含有杂原子取代的硅化合物(图6)。
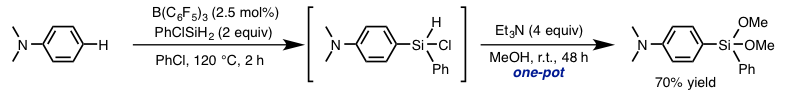
图 6. 杂原子取代的硅化合物的合成
总结
这一次我们介绍了不利于任何金属的硼催化的硅基化反应。该反应的优点虽然是显而易见的,但是并不能完全取代金属催化的硅基化反应。虽然该反应对硅基化试剂的适用性很广,但是由于底物限制在苯胺衍生物,这是该文章的一个弱点。
但是,对于C-H硅基化反应来说本文中利用正电性的硅活性中间体这一个方法是十分少见的,或许这将为以后的进一步的硼催化硅基化反应提供一扇大门。
参考文献
- Cheng, C.; Hartwig, J. F. Chem. Rev. 2015, 115, 8946. DOI: 10.1021/cr5006414
- Cheng, C.; Hartwig, J. F. Science 2014, 136, 12064. DOI: 10.1021/ja505844k
- Cheng, C.; Hartwig, J. F. J. Am. Chem. Soc. 2015, 137, 592. DOI: 10.1021/ja505844k
- Toutov, A. A.; Liu, W. B.; Betz, K. N.; Fedorov, A.; Stoltz, B. M.; Grubbs, R. H. Nature 2015, 518, 80. Di: 10.1038/nature14126
- (a) Blackwell, J. M.; Foster, K. L.; Beck, V. H.; Piers, W. E. J. Org. Chem. 1999, 64, 4887. DOI:10.1021/jo9903003(b) Parks, D. J.; Blackwell, M.; Piers, W. E. J. Org. Chem. 2000, 65, 3090. DOI:10.1021/jo991828a (c) Piers, W. E.; Marwitz, A. J. V.; Mercier, L. G. Inorg. Chem. 2011, 50, 12252. DOI: 10.1021/ic2006474 (d) Houghton, A. Y.; Hurmalainen, J.; Mansikkamaki, A.; Piers, W. E.; Tuononen, H. M. Nat. Chem. 2014, 6, 983. DOI: 10.1038/nchem.2063
- Ma, Y.; Wang, B.; Zhang, L.; Hou, Z. J. Am. Chem. Soc., 2016, 138, 3663. DOI:10.1021/jacs.6b01349
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.