
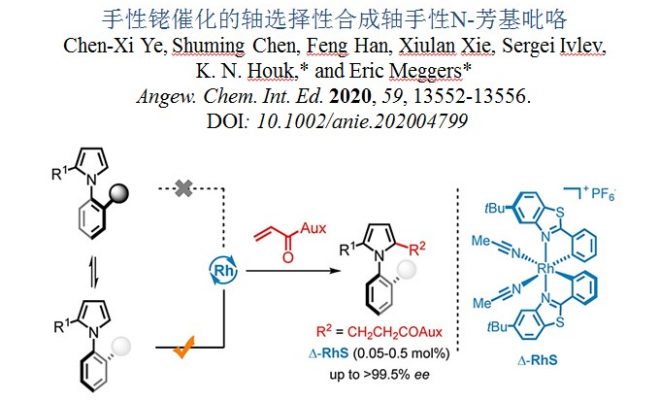
导读
轴手性联芳基化合物可作为手性试剂、催化剂、生物活性化合物以及功能材料,在有机化学中占据非常重要位置。[1]由于轴手性N-芳基吡咯化合物具有特殊的结构和电子性质,并且能够对富含电子的芳基吡咯部分直接进行功能化,因此,N-芳基吡咯化合物正日益受到有机化学家的广泛关注。目前,轴手性N-芳基吡咯可以作为拆分外消旋体的手性试剂以及手性配位配体、手性有机催化剂等。
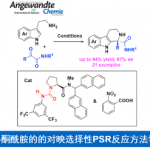
尽管轴手性N-芳基吡咯在有机合成中具有十分重要的地位,但是目前为止,获取轴手性N-芳基吡咯化合物的大多数方法均是利用外消旋体的手性拆分。[2]目前利用不对称催化策略构筑N-芳基吡咯化合物仅有两例,均为谭斌课题组发展的。其中一例利用苯胺和1,4-二酮为起始原料,在Lewis酸和手性磷酸(CPA)共同催化下发生Paal-Knorr反应,进而不对称合成N-芳基吡咯(图1a)。[3]然而该反应中催化剂用量高,反应速率慢且底物范围有限。另外一例是利用前手性或外消旋2,5-二取代N-芳基吡咯类化合物为起始底物,在CPA催化下利用去对称化或动力学拆分策略构筑高对映选择性的轴手性N-芳基吡咯(图1b),但该工作中需要在3位或4位引入大体积的丙二酸酮,仍具有一定局限性。[4]基于以上研究背景,德国马尔堡大学化学系Eric Meggers教授发展了一例手性铑催化的高对映选择性合成N-芳基吡咯。该方法利用了手性金属铑催化剂独特的螺旋手性,提供了一个构筑结构多样的轴手性N-芳基吡咯的新方法。相关成果发表于《Angew. Chem. Int. Ed. 》上:
“Atroposelective Synthesis of Axially Chiral N-Arylpyrroles by Chiralat-Rhodium Catalysis”
Chen-Xi Ye, Shuming Chen, Feng Han, XiulanXie, Sergei Ivlev, K. N. Houk,* and Eric Meggers*
Angew. Chem. Int. Ed. 2020, 59, 13552-13556. DOI: 10.1002/anie.202004799
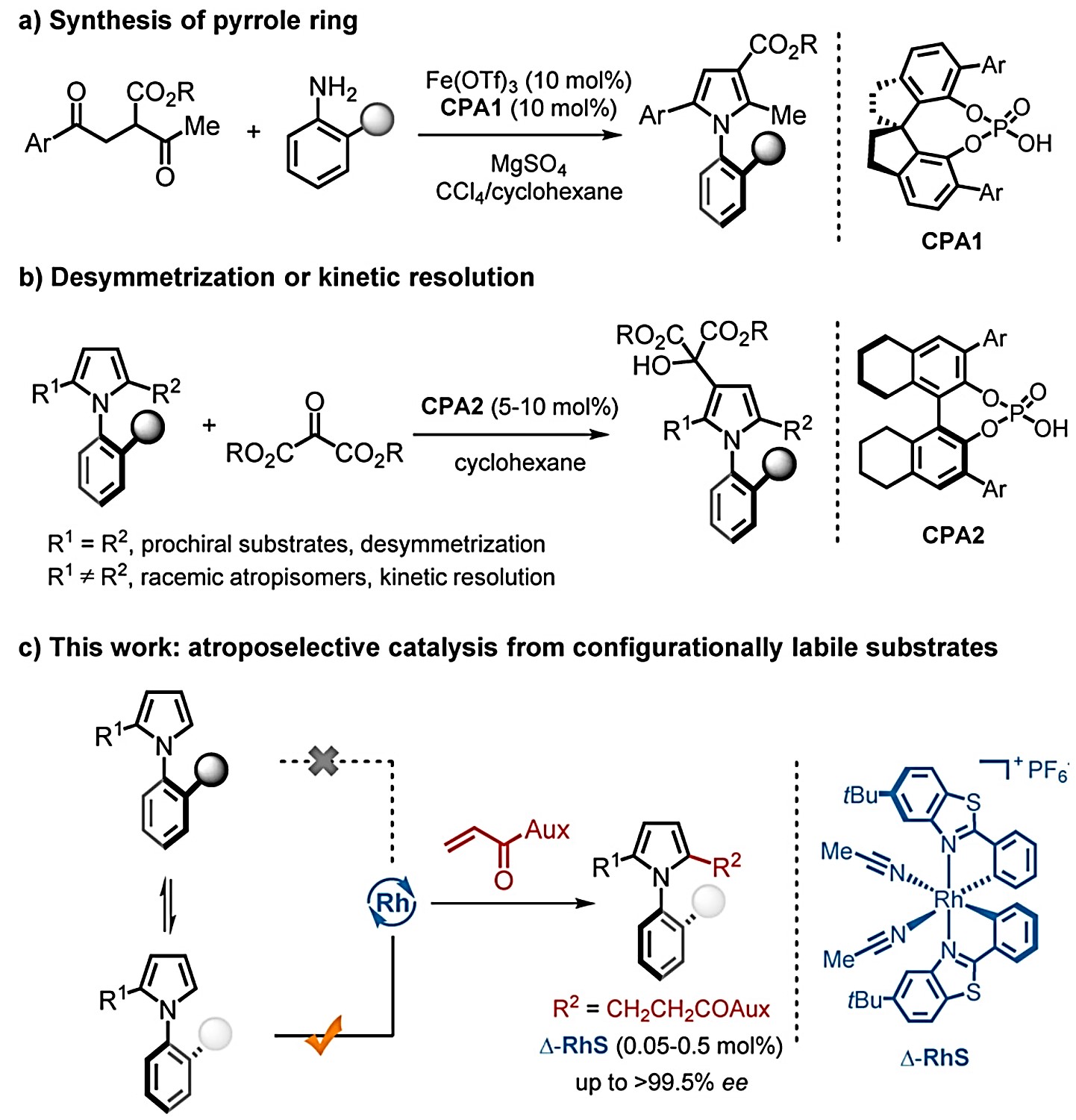
图1.不对称合成轴手性N-芳基吡咯类化合物
论文概要
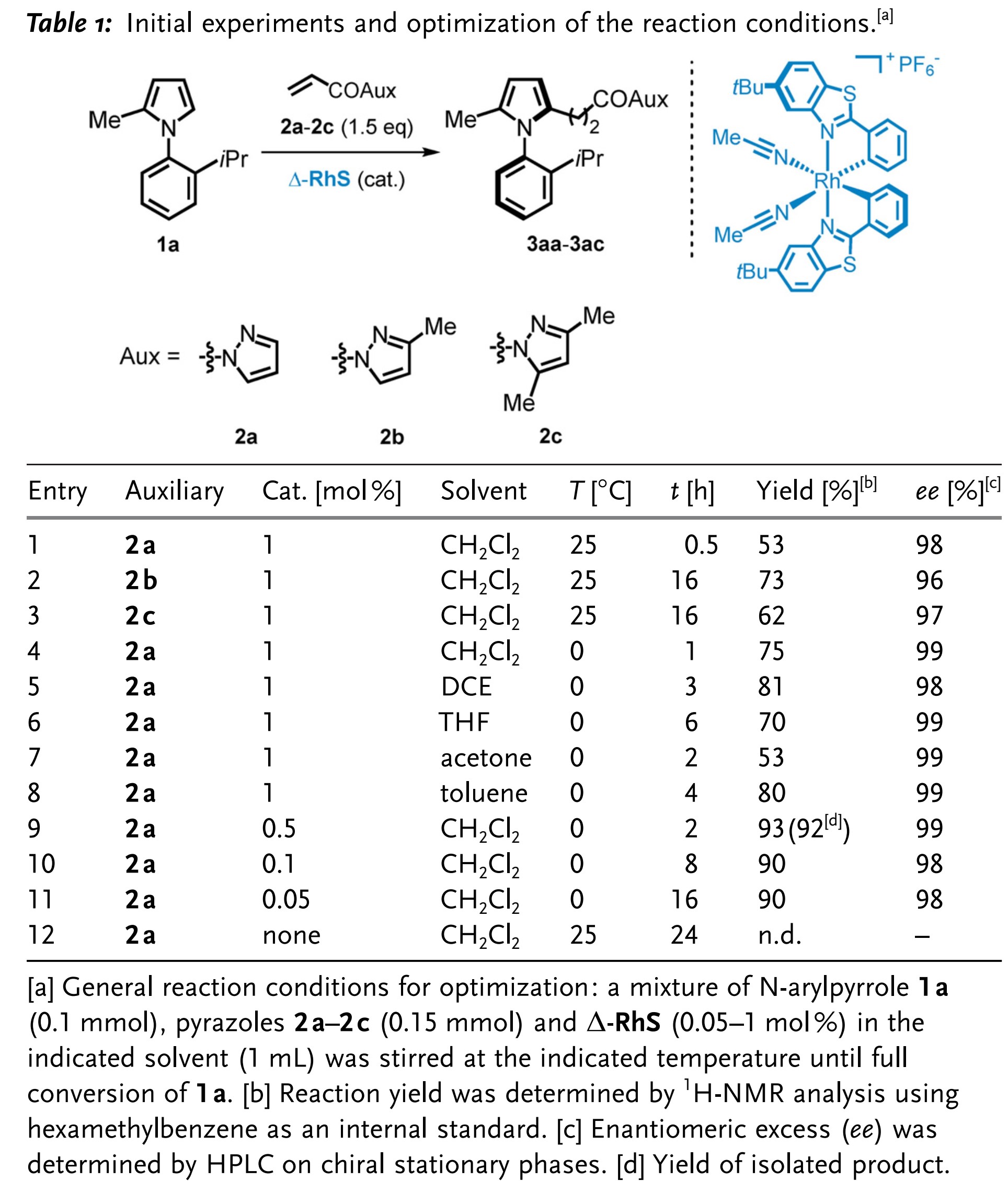
作者以N-(2-异丙基苯基)-2-甲基吡咯(1a)为模型底物开展研究。该底物具有轴手性,但在室温下,两个异构体迅速相互转化。作者首先在1 mol%双环金属铑催化剂Δ-RhS的催化下,N-芳基吡咯1a与N-丙烯酰-1H-吡唑(2a)可在室温下进行反应,以53%的收率以及98%的优异对映选择性得到目标产物3aa(表1,entry 1)。作者认为造成收率较低的原因为生成了其他副产物,即底物2a的吡咯环有多重取代反应发生。在相同反应条件下,甲基取代吡唑类底物2b和2c反应较慢,且ee值略微降低,但是收率略微提高,作者认为,取代基可能增大底物位阻,抑制副产物的生成(表1,entry 2 & 3)。当反应温度降低到0 ℃时,反应时间略微加长,但是目标产物的收率和ee值均有所提高(表1,entry 4)。接下来作者对溶剂展开研究,并发现该反应对溶剂的耐受范围很广,不同溶剂下的目标产物均具有优异的ee值(表1,entry 5-8)。鉴于在CH2Cl2中进行反应速率最高,因此作者选择CH2Cl2为最佳溶剂。此外,作者也对催化剂的用量进行研究。实验表明,0.5mol%的Δ-RhS催化剂用量为最佳(表1,entry 9)。此外,催化剂的用量可降低至0.05 mol%,也能在合理反应时间内保证反应的收率和对映选择性。在没有Δ-RhS的情况下,该反应并不能发生。因此,作者最终选定0.5mol%的Δ-RhS催化剂用量、CH2Cl2为最佳溶剂、在0 ℃下进行反应为最佳反应条件。
表1反应条件筛选
在确定了最佳反应条件后,作者进行了底物拓展研究。首先对N-芳香吡咯类化合物的吡咯环上的不同取代基进行了研究(图2)。对于不同的2-取代吡咯,反应均能以高收率以及高对映选择性(高达99% ee)得到目标产物3aa-3d,并且可以观察到较大位阻取代基对应的产物ee值有所降低。当2,3-二烷基吡咯类进行反应时,反应仅能以中等的收率得到目标产物(3e&3f),这可能因为多取代基使吡咯环上电子密度的增加,加速了多取代副反应的发生,从而使单取代产物的收率降低。
作者也对N-芳基吡咯的苯环上不同的取代基进行了拓展研究,发现反应均能以高收率以及优异对映选择性(高达>99% ee)得到目标产物3g-3s。值得一提的是,底物1i具有较大位阻的叔丁基,因此反应以动力学拆分过程进行,并且当底物1i的用量为2.5当量时,该反应也能以很好的收率(91%)以及对映选择性(>99% ee)得到目标产物3i。具有多环取代的底物也能很好的适应该反应,能以很高的收率以及对映选择性得到目标产物3k、3l、3m。此外,卤素取代基也能很好的应用于该反应中,以高收率和优异对映选择性得到目标产物3o-3q,也为后续的更多偶联反应提供了可能性。苯环上具有未保护的氨基也可以进行反应,得到目标产物3r,并有可能作为手性有机催化剂使用。含有羟基、硝基的特殊底物也能很好的适应该反应,得到目标产物3n和3s。值得一提的是,产物3ad同时具有碳中心手性以及轴手性,且有很高的立体选择性(>20:1dr和>99.5%ee)。
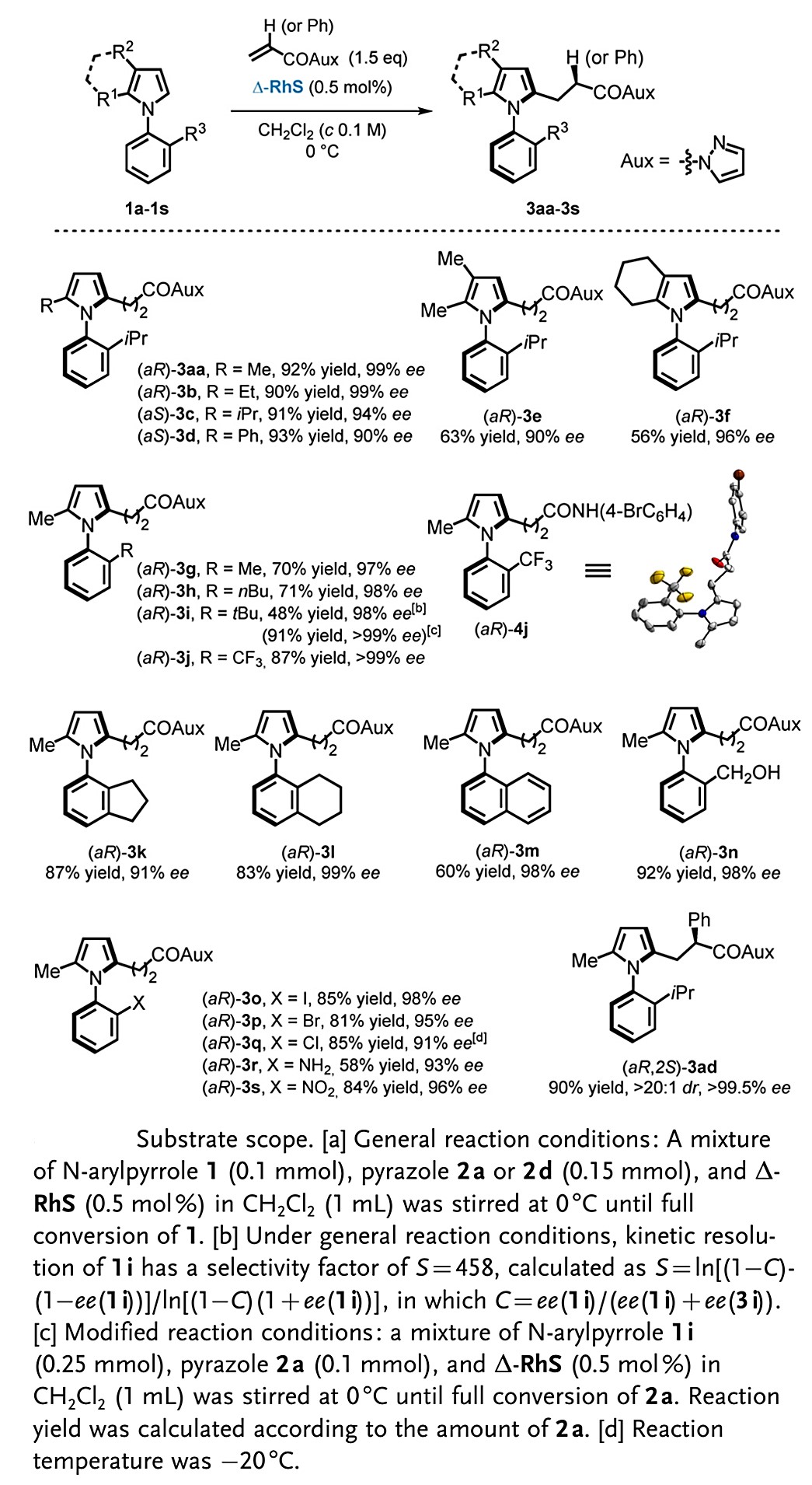
图2底物拓展研究
作者也对该反应进行了密度泛函理论(DFT)的计算,以了解该反应的立体选择性的起源(图3)。N-芳基吡咯化合物是可以瞬时、相互转化的异构体,能够迅速外消旋化。Δ-RhS活化的丙烯酰吡唑能与两种异构体发生反应,(aS)-N-芳基吡咯(TS-1a)的反应能垒比(aR)-萘基吡咯(TS-1b)的反应能垒低2.1 kcal/mol。在TS-1b中,N-芳基吡咯上的异丙基与配体的叔丁基存在立体排斥(最近的H···H距离2.35Å),在TS-1a中没有不稳定的相互作用。
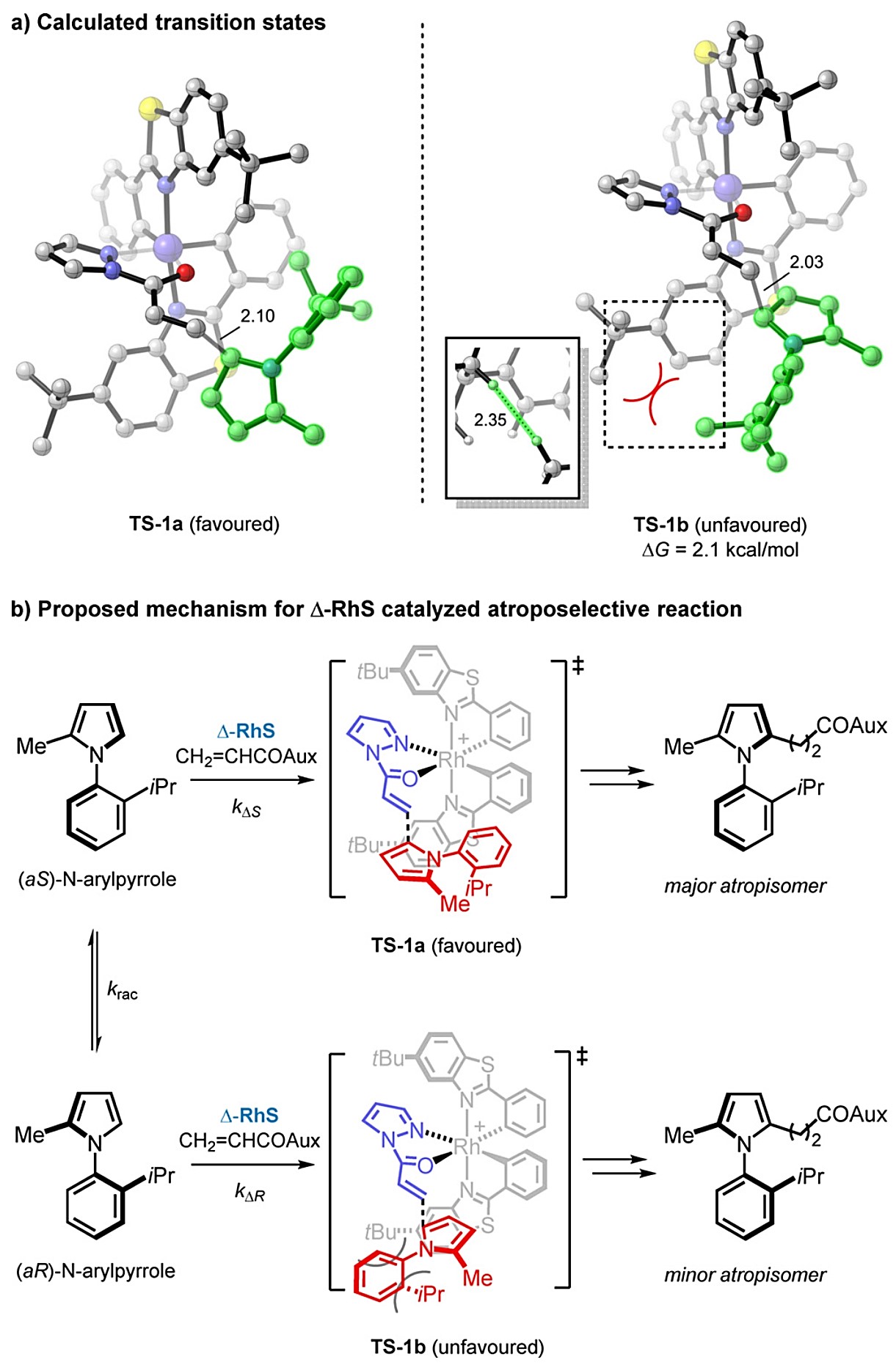
图3 密度泛函理论(DFT)计算研究
作者为了证明该反应的应用价值,对3o进行了克级合成。在标准条件下,能以88%的收率以及98%的ee值得到3o。此外,作者采用3o作为底物,进行了更多的衍生化尝试(图4)。此外,N-芳基吡唑也可以通过使用适当的还原剂进行还原。例如,NaBH4可将3o还原为一级醇7o,并能在钯催化条件下,转化成具有膦取代的N-芳基吡咯8o。使用DIBAL-H作为还原剂,可以将7o还原为醛9o,醛9o作为理想的底物可以进一步转化为乙烯基吡咯10o。
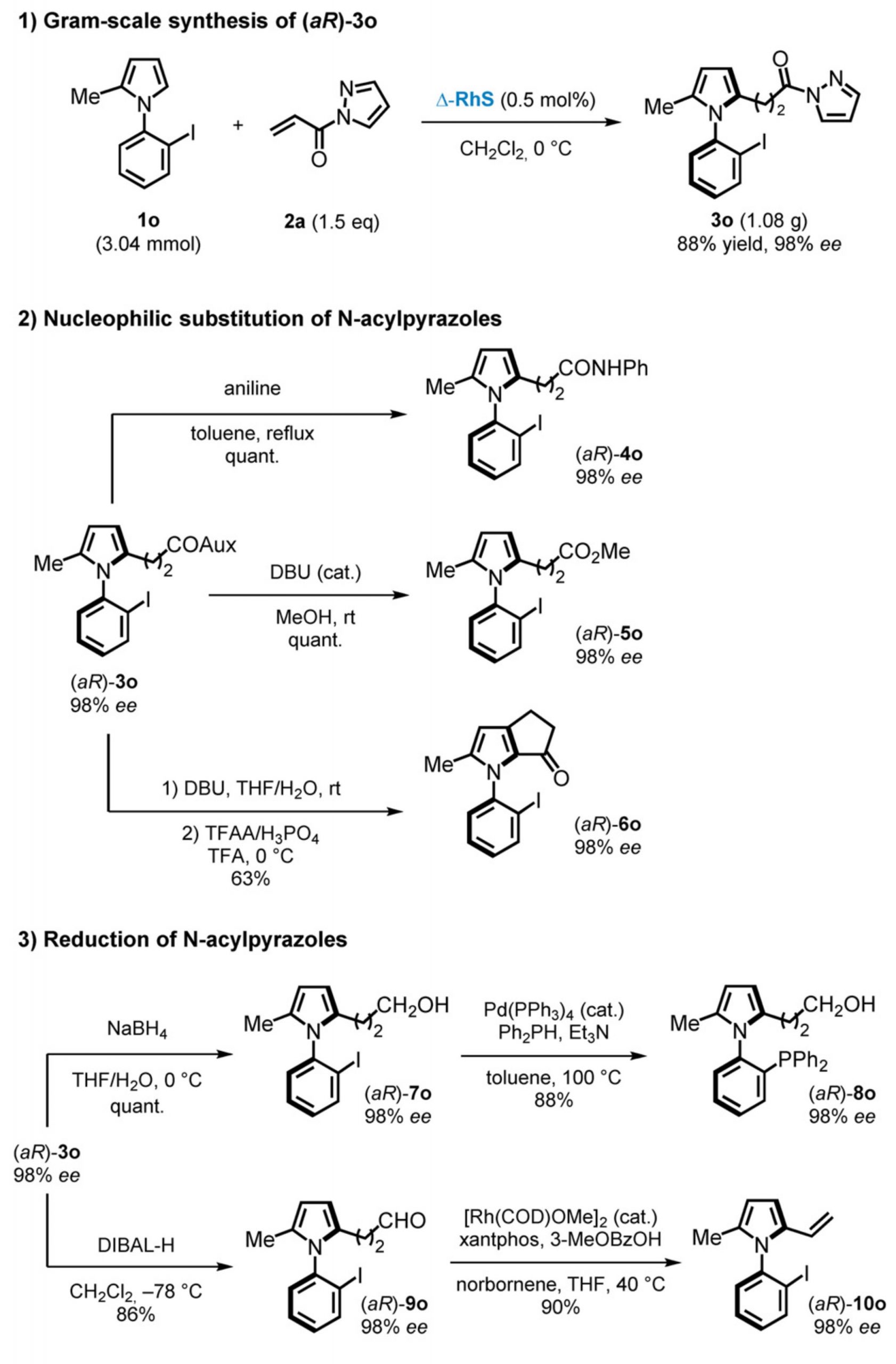
图4衍生化反应研究
论文总结评价
德国马尔堡大学化学系Eric Meggers教授发展了一例手性铑催化的高对映选择性合成轴手性N-芳基吡咯类化合物的反应,且该反应具有优良的立体选择性以及底物适应性。烷基化的轴手性N-芳基吡咯能够通过衍生化反应实现结构的多样性转化。此外,利用DFT计算表征了反应过渡态并揭示了反应的立体选择性起源,为未来相关反应的研究奠定了基础。
参考文献
- [1] Bringmann, T. Gulder, T. A. M. Gulder, M. Breuning, Chem.Rev.,2011, 111, 563-639.DOI:10.1021/cr100155e
- [2] Faigl, B. Mátravölgyi, T. Holczbauer, M. Czugler, J. Madarász,Tetrahedron: Asymmetry,2011, 22, 1879-1884. DOI: 10.1016/j.tetasy.2011.10.021
- [3] Zhang, J. Zhang, J. Ma, D.-J. Cheng, B. Tan, J. Am. Chem. Soc., 2017, 139, 1714-1717. DOI: 10.1021/jacs.6b09634
- [4] Zhang, S.-H. Xiang, J. Wang, J. Xiao, J.-Q. Wang, B. Tan,Nat.Commun.,2019, 10, 566. DOI:10.1038/s41467-019-08447-z
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
















No comments yet.