
本文作者:杉杉
导读
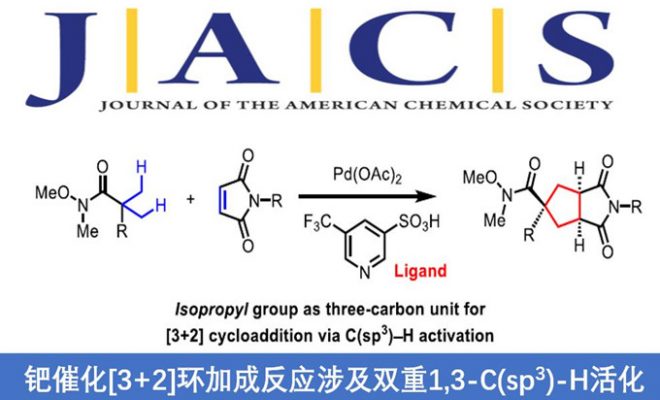
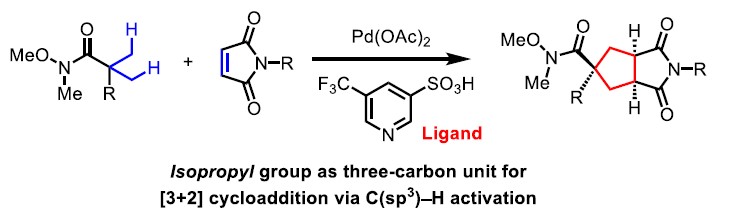
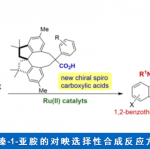
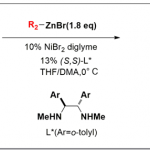
环加成反应作为快速构建环状化合物的一种快捷途径,然而,典型的环加成反应常需在底物内引入多个反应活性基团(如π-键,离去基团等),从而导致反应效率低或范围窄的问题。近日,美国Scripps研究所余金权教授课题组在J. Am. Chem. Soc.发表论文,报道了钯催化的[3+2]环加成反应,该反应涉及两次C(sp3)-H活化的过程,从而构建了环加成反应所需的三碳单元。首先,脂肪族酰胺的β-C(sp3)-H被活化,再经马来酰亚胺的插入,从而引发二次C(sp3)-H活化。成功的关键之处在于,使用弱配位酰胺作为导向基团。若使用较强配位的导向基团与马来酰亚胺偶联时,优先进行Heck或烷基化反应。为了促进酰胺导向的C(sp3)-H活化,使用3-吡啶磺酸(pyridine-3-sulfonic acid)配体至关重要。同时,该方法可与多种酰胺底物兼容,获得螺双环产物。而产物经还原性去对称化过程,能以优异的对映选择性获得具有多个立体中心的手性环戊烷类化合物。
Palladium-Catalyzed [3+2] Cycloaddition via Two-Fold 1,3-C(sp3)-H Activation
Hojoon Park and Jin-Quan Yu*
J. Am. Chem. Soc. 2020, 142, 16552. DOI:10.1021/jacs.0c08290
正文
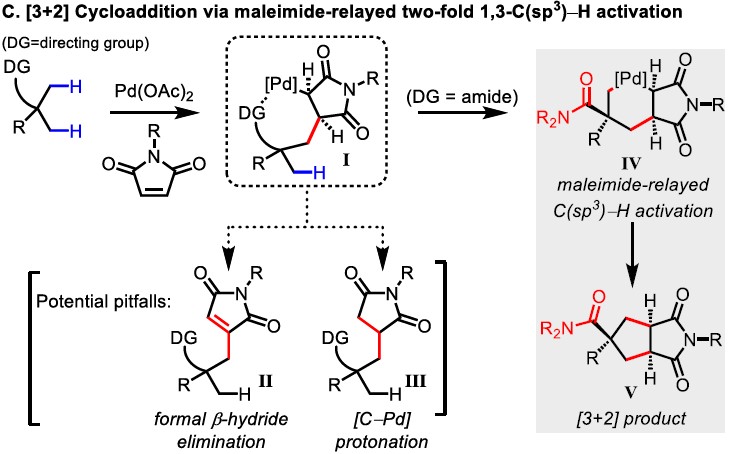
通过简单的方法构建环状化合物在有机合成中非常重要,而环加成反应作为此类反应代表,如[4+2] Diels-Alder反应。虽然[3+2]Diels-Alder反应可用于合成环戊烷衍生物,但选择合适的三碳单元以及控制其与环加成反应的反应活性极具挑战(Scheme 1A)。实际上,直接生成游离的三亚甲基甲烷(TMM)中间体作为三碳单元的[3+2]反应,需要专门底物的设计,其适用性有限。此外,已经开发出通过过渡金属催化产生TMM的策略,如Trost课题组将Pd-TMM配合物用作各种[3+2]反应的中间体,并进一步用于合成手性环戊烷。然而,这些TMM-等价物中间体的产生,仍然需要对前体进行多次预官能化。虽然还开发了用于环戊烷合成的其他优美的环加成方法,但需要在底物中引入多个π-键或离去基团。考虑到复杂的天然产物和生物活性分子中五元环的丰度,若以简单且丰富的底物(如羧酸或酰胺),直接实现[3+2]环加成,则非常具有吸引力。
在Pd催化的各种C-H功能化反应中,可将惰性C-H键直接转换为通用C-Pd键从而进行各类转化。在这方面,作者猜想是否可以使用C(sp3)-H活化来将简单且未官能化的三碳丙烷骨架用于[3+2]环加成反应,其中偕二甲基(gem-dimethyl)部分的C(sp3)-H键与双键直接环化(Scheme 1B)。此外,受降冰片烯还原的1,2-C(sp2)-H活化的启发,作者设想可以利用类似的烯烃取代的1,3-C(sp3)-H的两次活化过程,来实现所需的[3+2]环加成。然而,该过程涉及具有C(sp2)-H键有利的五元环钯,但对于具有惰性的1,3-C(sp3)-H键的活化极具挑战。
为了验证假设,作者将马来酰亚胺作为中继烯烃而不是降冰片烯。首先,与降冰片烯得到的产物相比,马来酰亚胺得到的产物具有显著的合成效用。其次,马来酰亚胺是反应性环状烯烃,已被用作Pd催化的C(sp3)-H官能化的偶联底物。但是,插入关键的马来酰亚胺的中间体(I)最好避免β-氢消除(Heck产物II)或质子化(烷基化产物III)途径(Scheme 1C)。据文献报道,新戊酸和马来酰亚胺通过β-氢消除可形成Heck产物。此外,通过[C(sp3)-Pd]质子化途径形成的烷基化产物与强配位的双齿导向基团有利。这些结果表明,可通过采用新的导向基团或配体来改变马来酰亚胺插入形成配合物(I)的途径。
作者推测,使用弱配位的导向基团可能会促进中继的C(sp3)-H活化,由于从导向基团中释放Pd中心更容易,这对于Pd在二次C(sp3)-H活化必不可少。基于本课题组开发的吡啶磺酸配体,使该反应能够利用酰胺的弱配位羰基作为导向基团。令人高兴的是,使用酰胺底物可能获得[3+2]产物(V),大概是涉及双烷钯环中间体(IV)。在此,美国Scripps研究所余金权教授课题组报道了,在钯催化下,通过双重C(sp3)-H活化的过程实现酰胺底物和马来酰亚胺之间的[3+2]环加成反应。
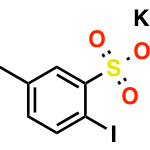
首先,作者以新戊酰胺1与N-(4-硝基苯基)马来酰亚胺作为模型底物,对配体进行了筛选(Table 1)。在无配体时,仅观察到10%收率的环戊烷产物以及痕量的Heck产物。当使用N-乙酰基甘氨酸(L1)或吡啶酮(L2)配体时,可改善结果,但使用吡啶-3-磺酸(L3)作为配体可显着提高产率。根据先前在Pd催化的C(sp3)-H烯基化反应中得到的结果,缺电子的L4是[3+2]反应的最佳配体。此外,在所有情况下,仅观察到Heck产物为次要产物,表明不管配体的性质如何,[3+2]途径是主要的。值得注意的是,当新戊酸与L4经历相同的[3+2]反应条件时,仅观察到Heck产物,这表明使用弱配位酰胺导向基团对于实现[3+2 ]途径至关重要。

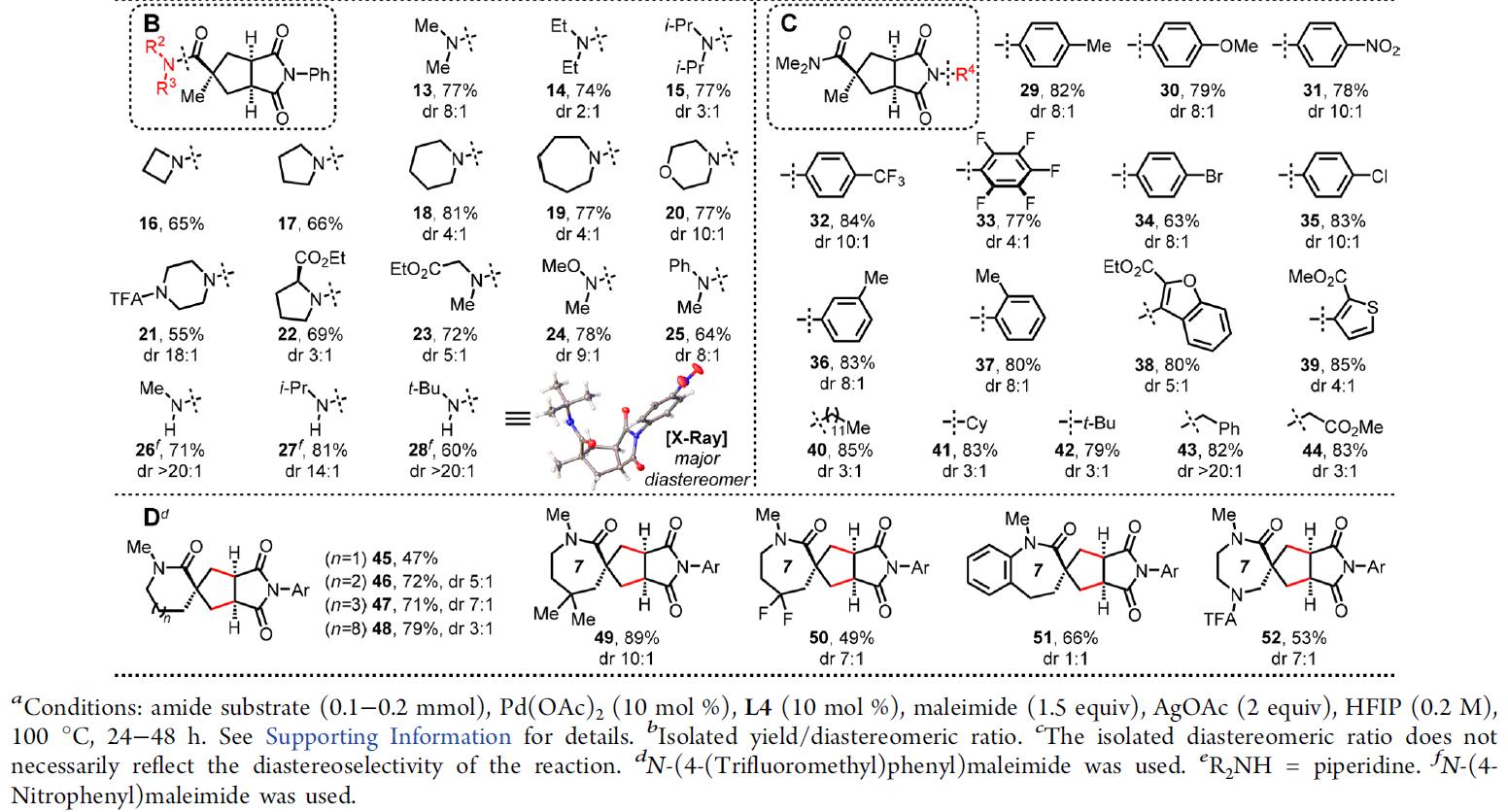
在获得上述最佳反应条件后,作者开始对酰胺底物和马来酰亚胺进行扩展(Table 2)。在对酰胺羧基上取代基研究结果表明(Table 2A),简单的烷基(2–3)、芳基(4)和含杂原子的官能团(5–8)都可得到所需的产物。含有饱和杂环(9–12)的底物也是合适底物。然而,带有α-氢的底物不兼容。
而对酰胺氨基上的底物范围研究表明(Table 2B),带有二烷基氨基(具有不同的空间位阻(13–15)和环状氨基(16–21))的底物,均可获得相应的产物。氨基酸衍生的底物(22–23)也获得很高的收率,Weinreb酰胺(24)和N-芳基(25)的底物同样兼容。虽然仲酰胺(26–28)也进行了所需的反应,但仍需缺电子的马来酰亚胺才能实现高收率。一般而言,简单的N-苯基马来酰亚胺与缺乏电子的马来酰亚胺相比,反应性较低,后者是活性更高的迈克尔受体,尤其是由于Thorpe-Ingold效应,与叔酰胺相比,仲酰胺的收率要低。
同时,在对马来酰亚胺N-取代底物研究表明(Table 2C),在N-芳基马来酰亚胺中,富电子的(29–30)和缺电子的(31–35)马来酰亚胺均具有高收率和非对映选择性。邻位和间位取代的N-芳基马来酰亚胺(36–37)也是合适的偶联底物。富电子的N-杂芳基马来酰亚胺(38–39)获得高收率的产物,但非对映选择性略低。N-烷基马来酰亚胺被证明是高反应性的偶联底物,无论N-取代基的性质如何(40–44)。然而,对于缺电子的烯烃(如马来酸酐),未获得目标产物。
最后,也将内酰胺作为[3+2]反应的底物进行研究(Table 2D),六元内酰胺(45)以47%的收率生成所需的双环产物,七元或更大的内酰胺(46–48)作为高反应性底物时,以高收率生成目标双环产物。功能化的七元内酰胺(49–51)以及1,4-二氮杂酮衍生的底物(52)也适用于[3+2]反应。

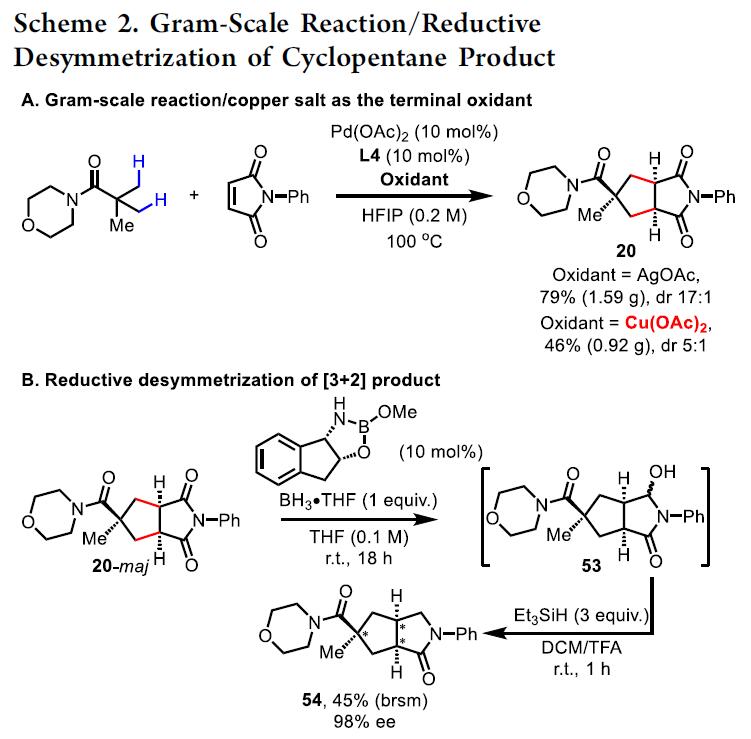
随后,为了进一步证明反应的实用性,作者进行了克级实验,不出所料,反应结果与小试一致(Scheme 3A)。当使用醋酸银或醋酸铜(廉价)作为末端氧化剂时,同样可以进行反应,但收率降低。同时,作者还尝试通过使用手性恶唑硼烷催化(3B)对产物进行还原去对称反应,从而获得手性环戊烷(Scheme 3B)。基于Jones课题组的研究,首先将[3+2]产物20用(1S,2R)-顺-1-氨基茚满-2-醇衍生的催化剂进行去对称反应,获得羟基内酰胺53。随后,将53直接转化为内酰胺54。令人高兴的是, 54的ee高达98%,尽管收率偏低(45%)。低收率是由于过度还原导致吡咯烷副产物的形成。然而,非对映选择性[3+2]环加成和对映选择性还原的组合,使其能够快速合成具有多个立体中心的新型手性环戊烷化合物。
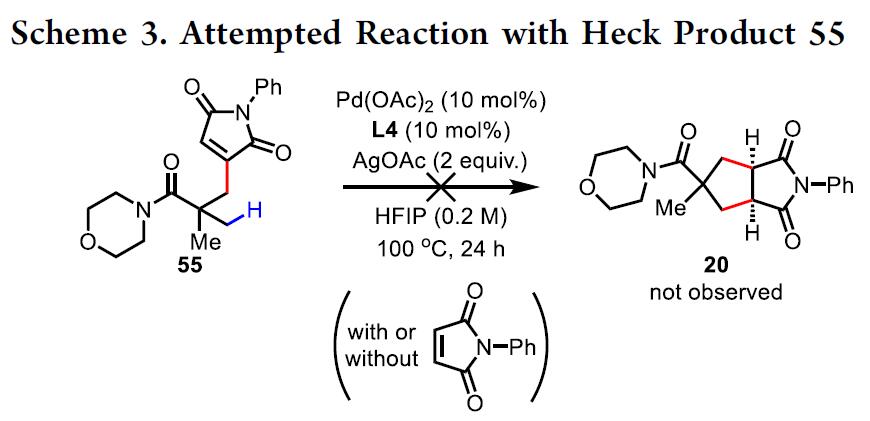
此外,该反应途径也可能先形成Heck产物,然后再发生第二个C(sp3)-H活化/分子内迁移。尽管由于[C-Pd]的加成反应,该过程将导致反式稠合的双环核,但热力学产物的快速差向异构化可能使其获得[3+2]产物。为了验证该途径的可行性,作者制备了Heck产物55,并在标准条件下反应(Scheme 4)。无论是否存在马来酰亚胺,都未观察到[3+2]产物20的形成,这意味着Heck产物不是[3+2]途径的中间体。
总结
美国Scripps研究所余金权教授课题组开发了一种通过钯催化的双重C(sp3)-H活化,实现酰胺底物和马来酰亚胺之间的 [3+2]环加成反应。弱配位酰胺导向基团对于[3+2]途径至关重要,因为先前已报道其他导向基团会产生Heck或烷基化产物。为了使这种酰胺定向的活化C(sp3)-H键,使用缺电子的3-吡啶磺酸配体至关重要。同时,以非对映选择性的方式获得了多种环戊烷产物,包括衍生自内酰胺底物的螺双环化合物。此外,使用手性恶唑硼烷催化,可将[3+2]产物进行去对称化,以极好的对映选择性得到手性环戊烷化合物。因此,鉴于两个C(sp3)-C(sp3)键都是直接由C(sp3)-H键形成而无需进行预功能化,该方法将作为合成常规环戊烷策略的补充。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!




















No comments yet.