
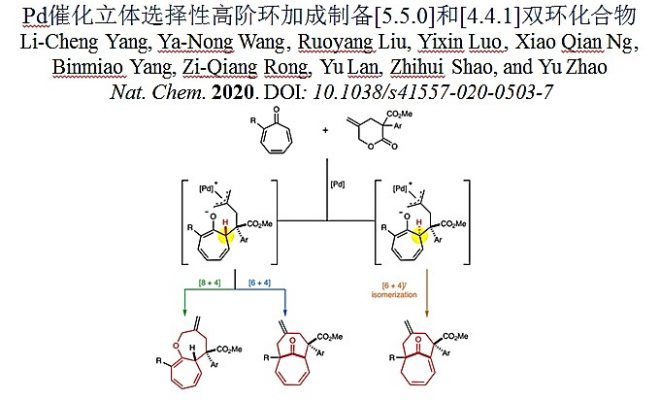
导读
中型环是许多天然产物的核心结构,在化学合成和药物合成中起着重要作用。特别是通常存在于生物活性分子中的并环或者桥联[5.5.0]和[4.4.1]双环化合物,如euglobal A(抗炎药),psiguadial A(抗人类免疫缺陷病毒),ingenol(抗癌)和环柠檬酸等。由于缺乏普遍、有效的化学合成中型环化合物的方法,尽管他们具有结构多样性和潜在的应用价值,但是在商业药品以及药物筛选中发展受限。此外,从共同的起始材料出发,通过不同的催化循环,立体选择性合成多种多环骨架化合物是非常吸引人的,且这样的转变可极大丰富化合物库,在药物开发方面具有重要意义。
在中型环合成的不同策略中,分子间高阶环加成在建立复杂多环结构方面具有显著优势。[1]以七元环酮为基础,特别是通过环庚三烯酮的环加成,进而立体选择性合成并环或桥联双环化合物的研究已经比较充分,然而,目前大多数的工作局限在五元或六元环的构筑。通过高阶环加成制备大型双环化合物的报导仍较少,目前仅有Jørgensen课题组报导的一例反应,即利用手性胺催化的环庚三烯酮对映选择性[6+4]环加成反应制备含[4.4.1]单元的化合物(图1a)。[2]此外,具有挑战性的并环[5.5.0]或含桥头烯烃[4.4.1]双环化合物,在复杂的天然产物合成中有很好的应用,报导的仅有分子内[5+2]环加成反应(图1b, c)。[3,4]这些报导需要使用设计合理的底物,且均以非立体选择性的方式进行合成。目前利用相应的分子间高阶环加成反应还未报导,更不用说利用同一催化体系、不同的途径开发多样性结构。作者一直致力于通过催化环加成反应立体选择性合成中型杂环化合物,在该工作中发展了钯催化环庚三烯酮的发散、立体选择性环加成反应,高效制备双环[5.5.0]十二碳三烯和桥环[4.4.1]十一烯,以及含桥头烯烃的[4.4.1]十一烯(图1d)。相关成果发表于:
“Stereoselective access to [5.5.0] and [4.4.1] bicyclic compounds through Pd-catalysed divergent higher-order cycloadditions”
Li-Cheng Yang, Ya-Nong Wang , Ruoyang Liu, Yixin Luo , Xiao Qian Ng , Binmiao Yang, Zi-Qiang Rong, Yu Lan, Zhihui Shao, and Yu Zhao
Nat. Chem. 2020. DOI: 10.1038/s41557-020-0503-7
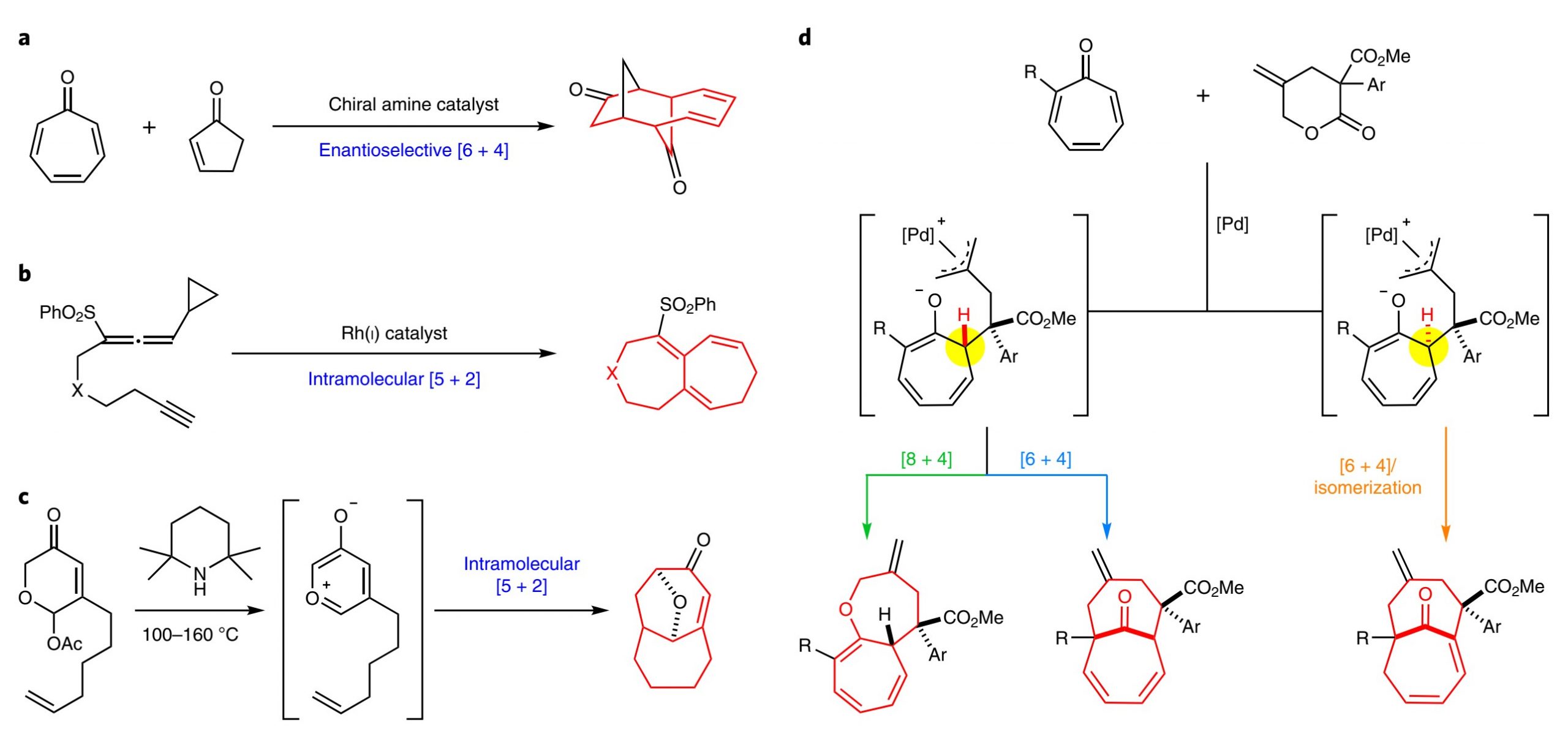
图1. 合成双环[5.5.0]以及桥环[4.4.1]类化合物
论文概要
条件筛选:作者首先进行反应条件的筛选,利用γ-亚甲基-δ-戊内酯2a为起始反应底物,和环庚三烯酮1a在钯催化下进行反应(图2a)。使用Pd2(dba)3⋅CHCl3和三异丙基亚磷酸酯(L1)进行催化,作者观察到了[6+4]环加合物4a和[8+4]环加合物3a,以及另一种含有桥头烯烃形式的[6+4]环加合物5a(使用L1)。尽管由于三种产品的分布不均(5:3:2)而导致的收率较低,但是三种产物均具有良好的非对映选择性(>20:1 d.r.)。因为这三种中型双环化合物产品都是有价值的,但很难实现立体选择性的合成(图1),因此,作者决定努力实现分别得到特定的目标化合物。作者研究了一系列金属前体和配体,如图2a所示。最终,通过筛选发现DBFphos(L2)对化合物3a的形成表现出最高的选择性(在-20 oC 88%的收率;条件A),同时dppf(L3)对4a的合成效果较好(81%的收率条件B);使用体积大的配体L4,通过温和加热,能以65%的收率得到5a(条件C)。此外,这三种产品均具有高收率以及非对映选择性(3a和4a的d.r.大于20:1;5a的d.r.为5:1)。
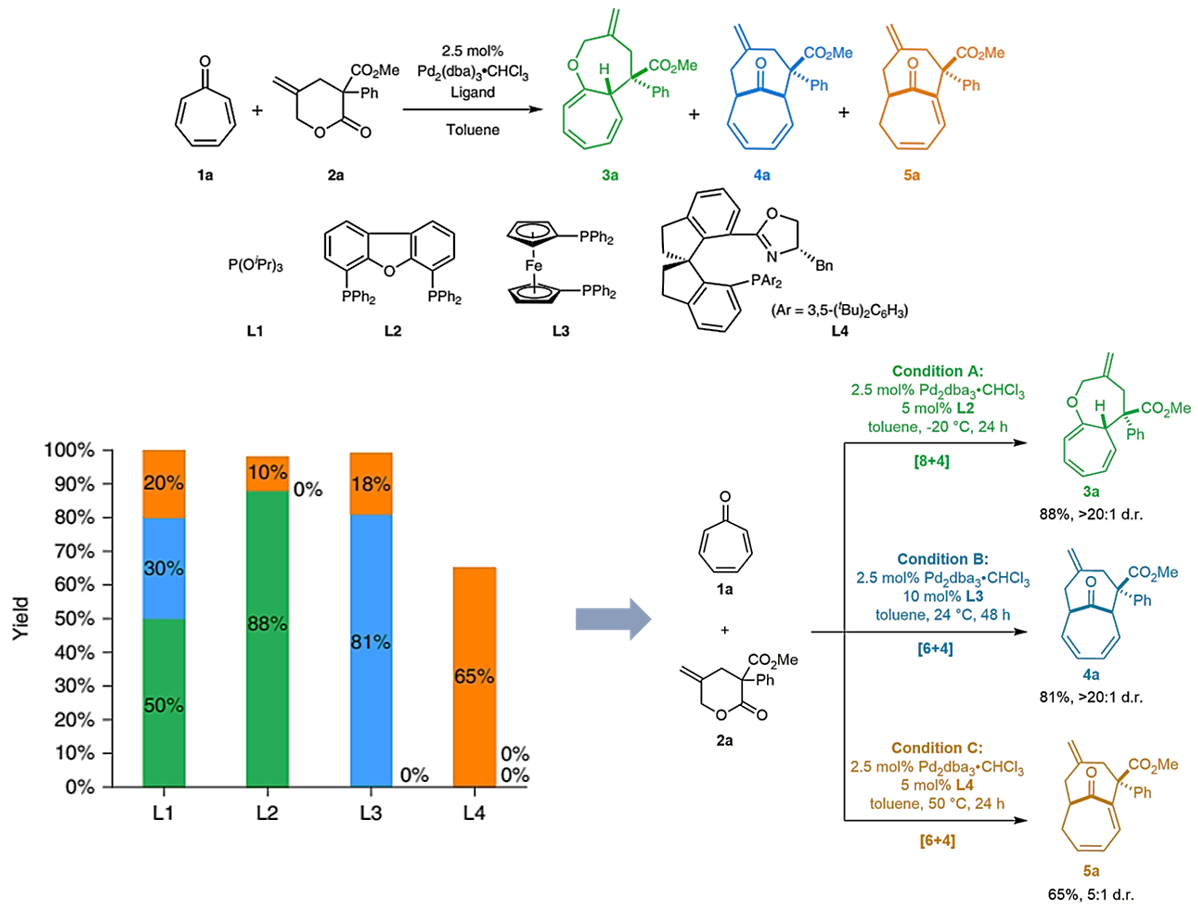
图2. 反应条件的建立
机理研究:为了探索该发散反应性的来源,作者对催化系统进行了详细的机理研究。首先,作者进行了控制实验,以4a的优化条件(条件B)进行研究。如图所示,在图3b中,底物环庚三烯酮1a被完全消耗,在最初的8小时内,伴随着主要产品3a(而不是4a)的形成,加上高达17%的5a。随着时间的推移,4a的逐渐形成,3a的减少,5a的量基本上保持不变。这些观察表明有两条不同的路径用于3a/4a和5a的形成。为了验证这个假设,作者接下来研究三种环加合物(图3c)。对于这项研究,与之密切相关的化合物3b、4b和5b含有相同的对甲氧基取代基。在B条件下,3b完全转化为4b(收率85%),与前面的反应一致。值得注意的是,将3b转换为4b需要钯催化剂继续催化,若没有催化剂,即使在100 oC的高温下未观察到转化。相反,4b到3b的反向转换完全没有发生。这些观察结果表明3b的形成是由于通过正常的[8+4]环加成,经过Pd催化还原O-烯丙基取代,然后C-烯丙基化,可以进一步转化为热力学上更稳定的[6+4]环加合物4b。进一步的研究表明, [6+4]环加合物5b不能与3b或4b相互转化,使其形成来自另一条催化路径。考虑到形成不同形式环加成反应的机理,我们假设3/4或5的反应性发散可能是由于形成了不同的非对映中间体。通过对与δ-戊二醛内酯的对甲基氧取代反应的探索,作者成功地观察到并分离出一个小的非对映体3b’。在相同的条件下(条件B),作者观察到3b’可以转化为5b,产率大于90%,而并没有观察到3b或4b(图3c)。这对最初形成的是非对映体中间产物提供了重要的证据,反应表现出明显的反应性差别来产生环加成产物3/4或含桥头烯烃5。为了阐明5的形成机制,我们制备了1a–d2,在两个α位置都有氘标记,并将其置于条件A和条件B下(图3d)。在条件A下,同时5a– d2和3a–d2型,在条件B下5a–d2和4a–d2。由此可见,3a–d2或4a–d2氘的位置在两个α位置不变。相比之下,5a–d2中的二烯转位,且氘从C1切换到C7。另外,氘在C7上,只在羰基的对面桥头(>20:1 d.r.)。这些关键的观察结果强烈地暗示了一种氢迁移机制。为了提供进一步的证据,作者用1a–d2与氯代1r进行交叉实验(图3e)。此外,未观察到交叉产物5,这也支持了分子内氢迁移机理。
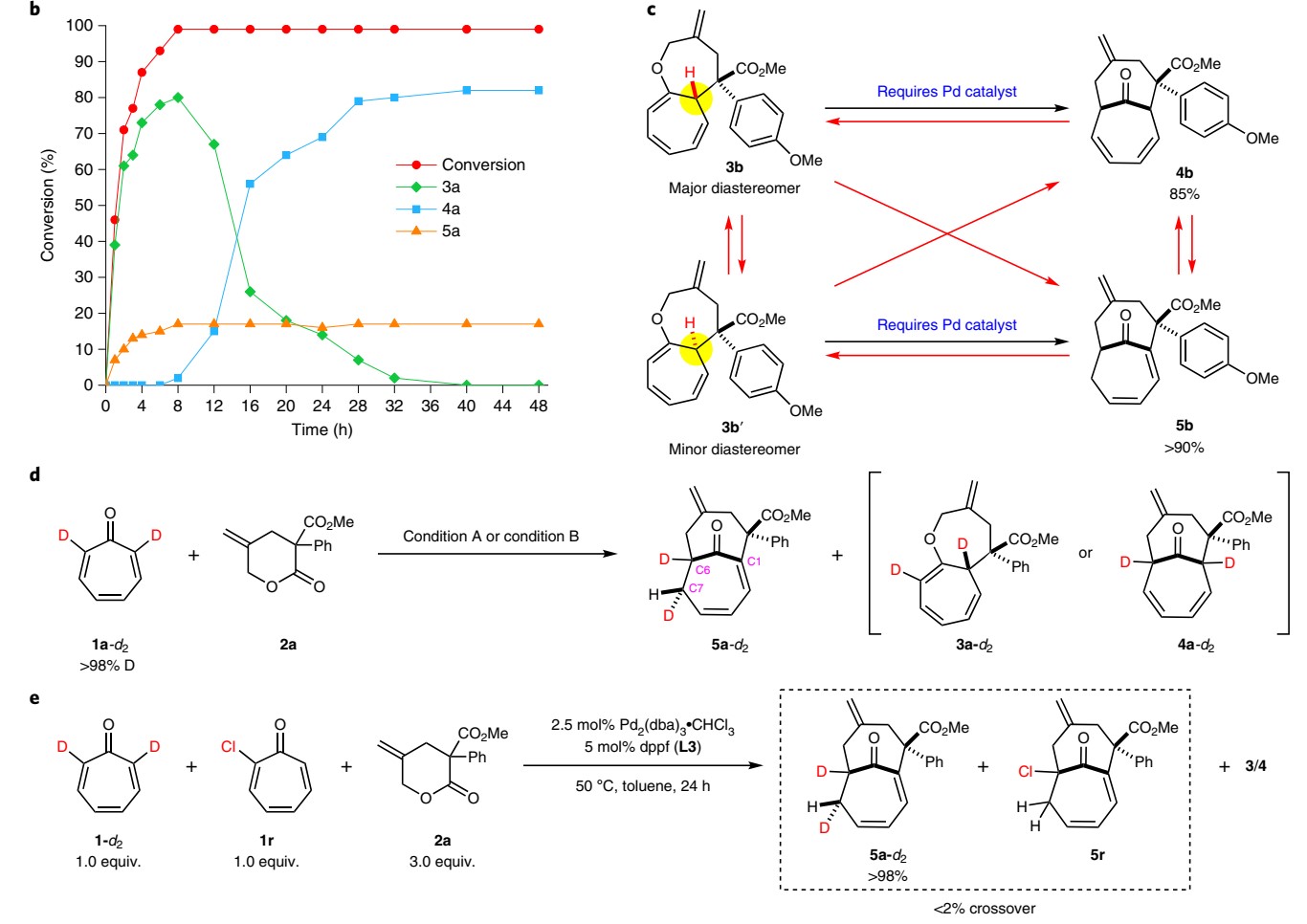
图3 控制实验
根据这些观察结果,作者提出的机理如图4a所示。在通过内酯2a的脱羧作用形成Pd-π-烯丙基Int-I后, Int-I碳负离子与环庚三烯酮1a发生分子间亲核攻击到,产生两个非对映体两性离子(Int II或Int III),可以进一步实现发散环加成的途径。为了得到更好的理解,作者将1a和2a在B条件下的模型反应进行DFT计算(使用M06-2X在甲苯溶剂中进行)。如图4b所示,在Int II(TS-II)的过渡态下,Int-I的苯基与1aα-H的扭转角是65.6o。TS-III中相应的扭转角由于环庚三烯酮羰基与芳基取代基上的C‒H之间的氢键相互作用,仅为10.7o。这会导致更大的排斥力,也就是说,TS-III的自由能更高(比TS-II高2.0 kcal /mol,形成Int II和Int II分别为21.4和23.4 kcal /mol)比
对于其他过程,因此,Int-I对环庚三烯酮的亲核攻击应该是速率决定步骤。此外,考虑到两个非对映体中间体是不可交换的,产生Int II或Int III的步骤应该是动力学控制,主要通过它们的能量差确定它们的比例。两性离子Int II中的O和C6都可以作为亲核试剂,与亲电钯-π-烯丙基部分反应。作者的观察和DFT计算均支持O-烯丙基化是最具动力学优势的,即生成[8+4]环加合物3a(via TS IV,
ΔG≠ = 3.8 kcal/ mol)。在钯催化下,该步骤是可逆的(ΔG≠ = 18.0 kcal/mol)。另一种C-烯丙基化可以提供热力学上更稳定的[6+4]环加合物4a(通过TS-V,ΔG≠ = 5.9k/cal)。此外,作者也对5的生成进行了探讨(图4),虽然作者在控制实验中初步证明了分子内氢迁移的机理,但是对于DFT计算来说,由于催化剂的不确定性,要确切证明生成5的异构化步骤过于复杂。
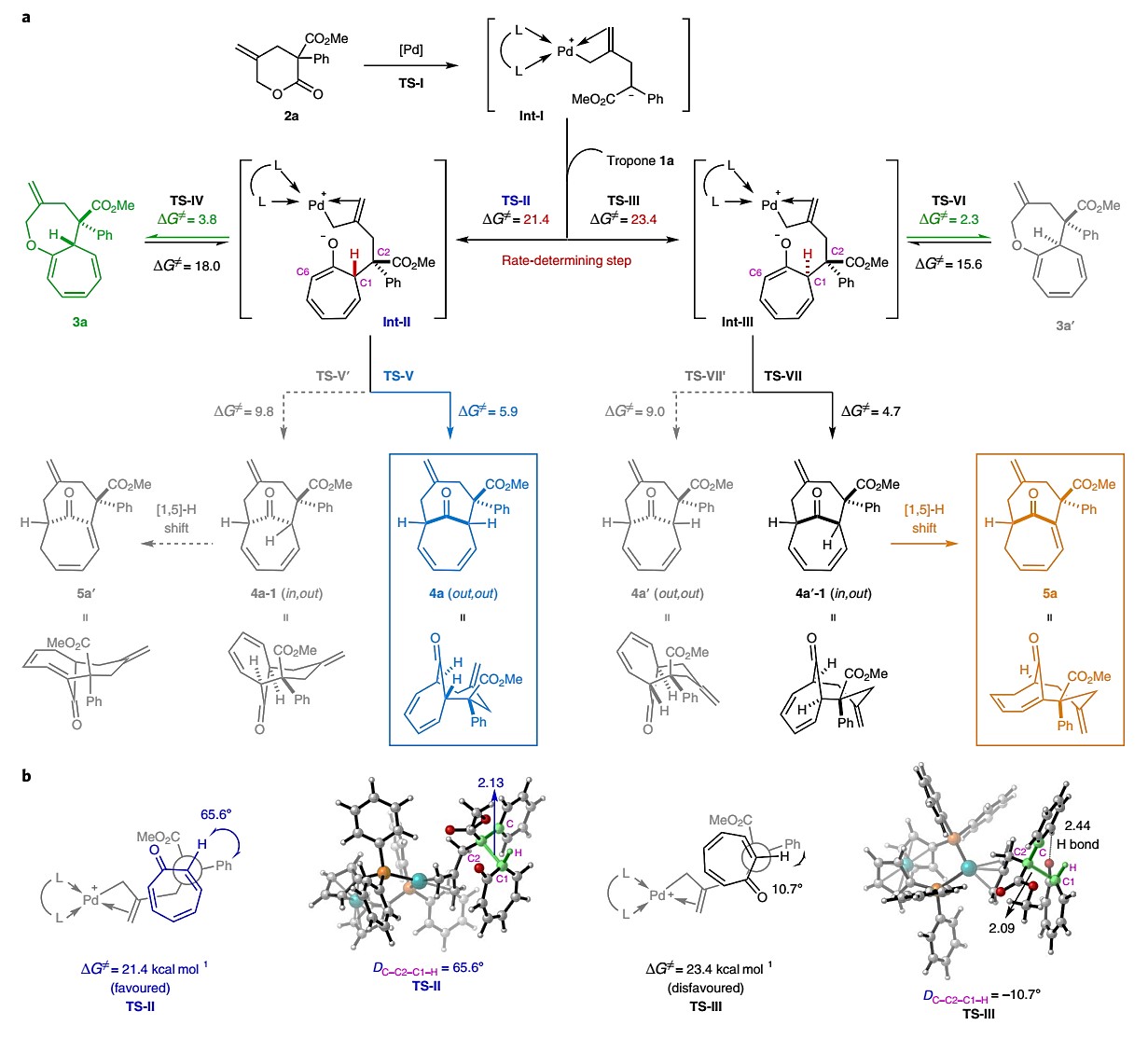
图4 DFT计算研究
底物拓展研究:作者对多种γ-亚甲基-δ-戊内酯以及环庚三烯酮均进行了研究,如表5所示。第一,各种含有对位和间取代芳基、杂环取代基以及叔丁酯,均可以在A或B条件下得到良好至优异收率和非对映选择性的双环[5.5.0]十二碳三烯3或双环[4.4.1]十一烯4(3a-3j和4a-4j)。2-乙酰氧基和叔丁氧基碳酸酯环庚三烯酮也可适用于该反应,得到相应的[4.4.1]环加成产物4k和4l,其收率分别为90%和75%,d.r.大于20:1。第二,为了得到含桥头烯烃的[4.4.1]双环5,各种含取代芳基的内酯2均可与环庚三烯酮在条件C下(使用L4)发生反应,得到5a-5c,和5f。出乎意料的是,当内酯2含有大位阻取代基时(例如,邻位取代的芳环或1-萘基),使用dppf(条件B)可生成5(而不是4)。此外,含杂环的2也可参与反应,以高收率和选择性生成吲哚衍生物5s。但是烷基取代的内酯2反应效果不佳,产物5t含苄基取代基收率低且选择性差。
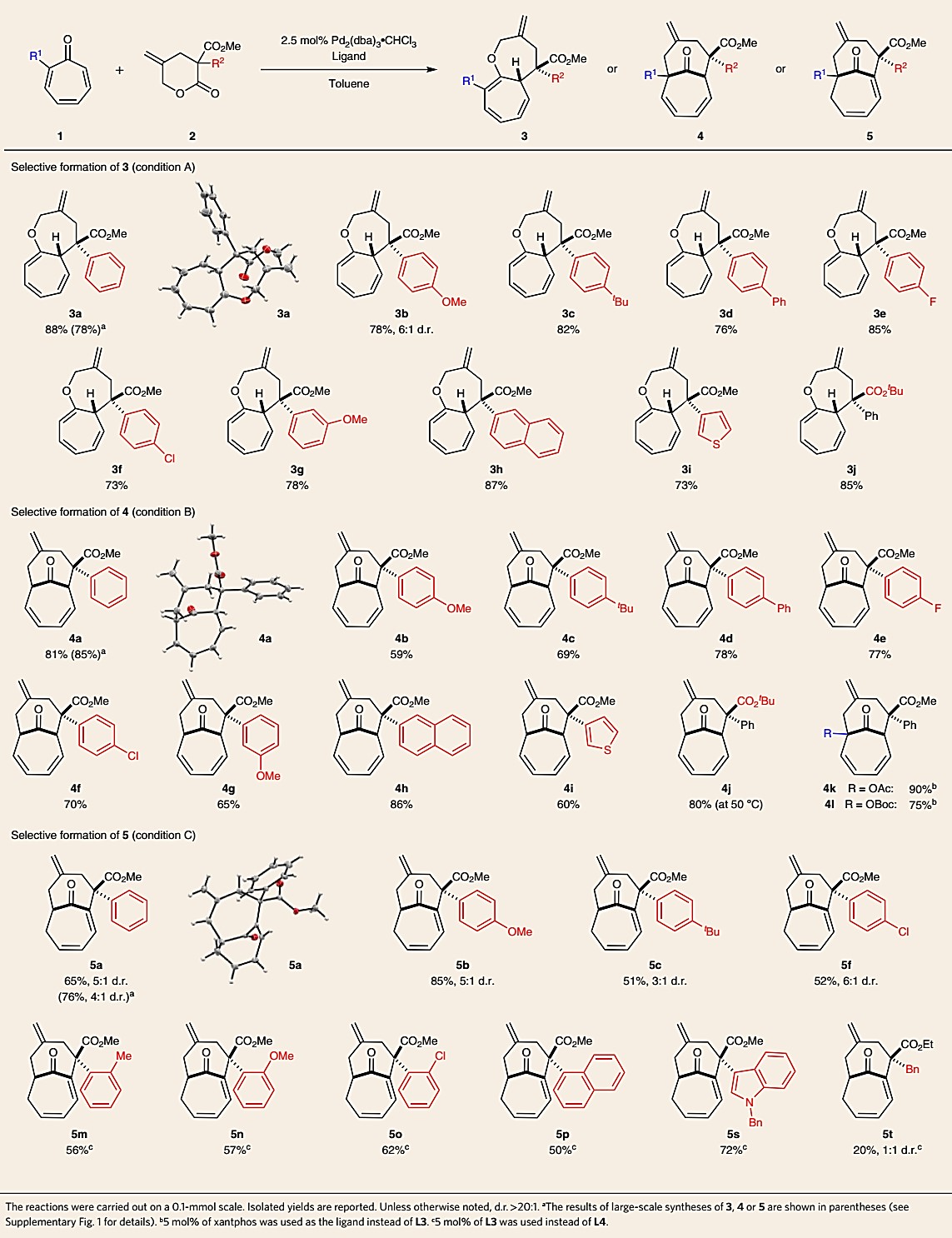
图5 不同条件下底物拓展研究
此外,作者在筛选出不同的手性配体后,对单膦配体(R)-SITCP参与的反应也进行了研究。在如图6所示的反应条件下,1a和2a可以发生反应,在温度为50 oC时,能一82-88% e.e.值得到目标产物4a和5a(图6),尽管比例接近1:1,但它们可以很容易地分离出来,收率分别为48%和42%。对于γ-亚甲基-δ-戊二醛内酯的变化,包括给电子和吸电子取代基,均能很好地适应于该反应,得到4(4b,4c和4f)和5(5b,5c和5f),收率适中,对映选择性好(80-90% e.e.)。此外,根据机理研究,动力学产物bicyclo[5.5.0]3在反应过程中生成。如图6所示,反应40min后停止,能分离得到3a (49%,82% e.e.)。此外,作者还考察环庚三烯酮的适应性。各种苯基、乙氧基、叔丁氧基碳酸酯和氯等均可以参与该环加成反应。有趣的是,这些反应对含有桥头烯烃双环5的有更高的选择性,均具有高收率和立体选择性(4:1-20:1 d.r.,80-88% e.e.)。作者进一步评价了该方法的适用范围,使用2-氯取代的环庚三烯酮进行反应,芳基上含有各种各样对位取代基的γ-亚甲基-δ-戊二醛内酯均能以良好的对映选择性(81-85% e.e.)生成5ra-5re。类似地,邻位取代芳基、2-萘基和噻吩的耐受性也较好,可产生5rf-5ri,且具有良好的立体选择性(>20:1 d.r.,67–85% e.e.)。通过一次重结晶,5q和5rh的对映选择性可进一步提高到大于99% e.e.。这一系列化合物的绝对构型由5rh的单晶X射线衍射分析得到。
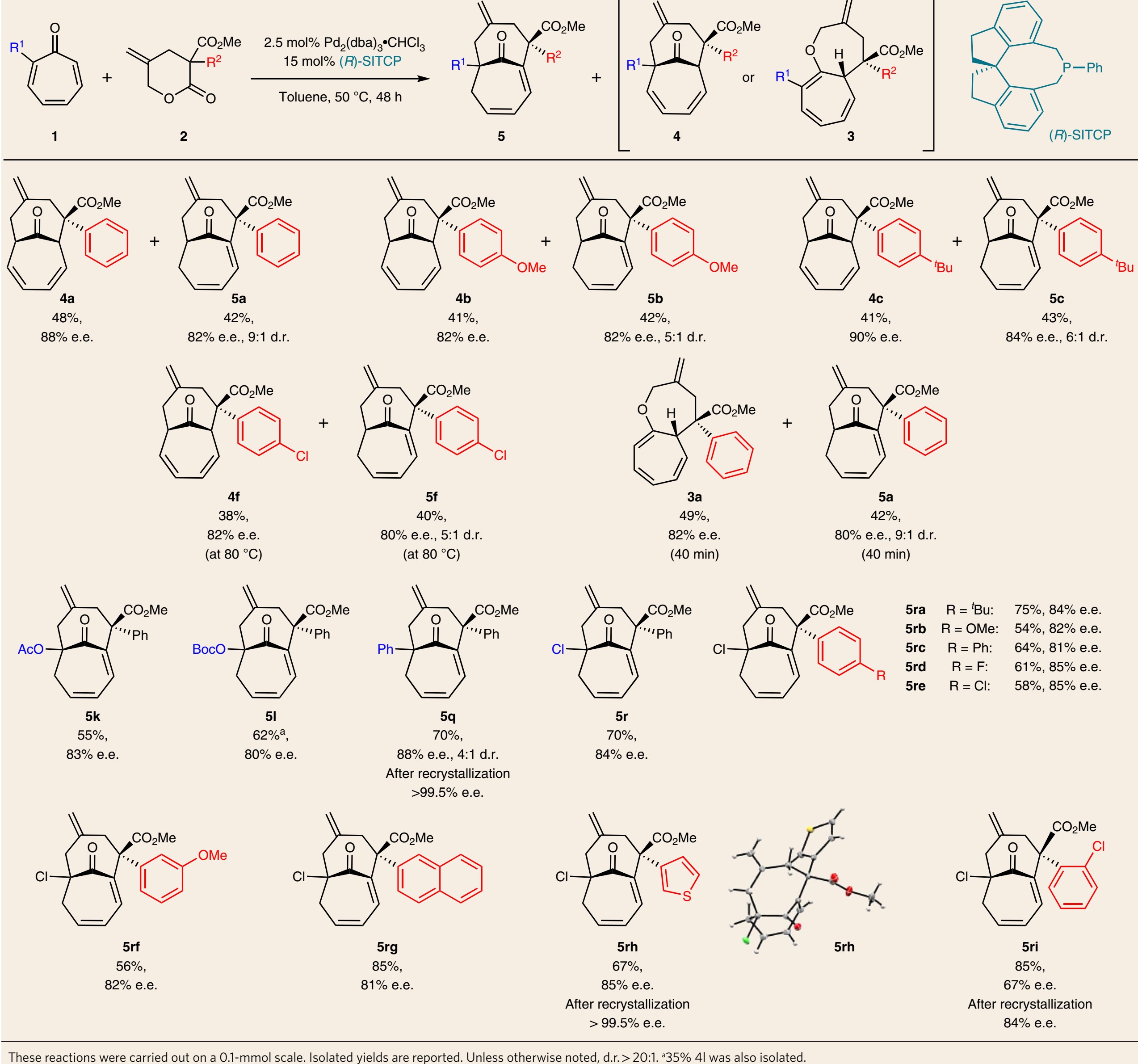
图6 其他底物拓展研究
衍生化反应:为了进一步展示该催化体系的效用,作者对化合物3-5进行了各种衍生化转化。如图7a所示,当用在原位生成苯炔时,反应得到新的双环化合物6a。由于这些桥联双环化合物的刚性构象,作者预期烯烃4、5的功能化的应该也是高度非对映选择性的。作者使用标准Pd/C系统对4a和5a进行氢化,7a和8a均以高的d.r.得到(图7b)。值得注意的是,8a中的桥头烯烃在此条件下保持完整。作者还尝试了4a和5a中桥羰基的立体选择性还原,如图7c所示,4a或5a中的羰基高度非对映选择性的还原如期发生,并进一步发生分子内酯化形成多环内酯9a和10a。类似的转变也可以发生在使用甲醇钠作为亲核试剂,以单对映体高收率得到11a和12a。
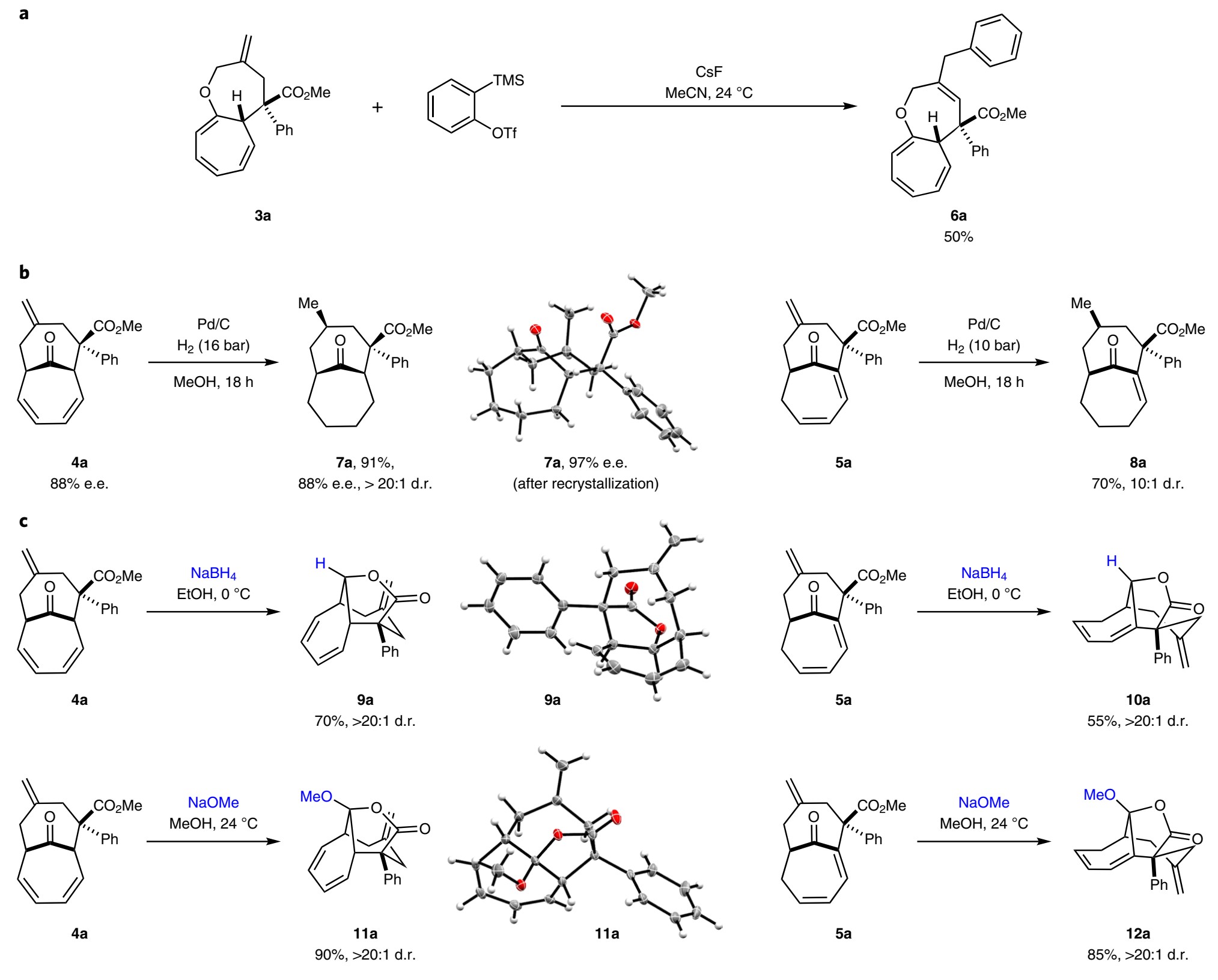
图7 可能的催化循环
论文总结评价
作者开发了一种高效的、发散的非对映选择性的合成方法,利用易得的环庚三烯酮和γ-亚甲基-δ-戊二醛内酯的高阶环加成反应,可以获得三类环化合物(双环[5.5.0]十二碳三烯,双环[4.4.1]十一烯以及含有桥头烯烃的[4.4.1]环)。作者也进行了详细的机理研究和DFT计算,解释了该催化体系中形成中间产物的发散反应性。作者也对这些中型双环化合物的进一步衍生化转化,以高非对映选择性高效得到一系列中等大小的双环和多环化合物。
参考文献
- [1] H. Rigby, Acc. Chem. Res. 1993, 26, 579-585. DOI: 10.1021/ar00035a003
- [2] Yu, Cyndi Q. He, A. Simon, W. Li, R. Mose, M. K. Thøgersen, K. A. Jørgensen, and K. N. Houk* J. Am. Chem. Soc. 2018, 140, 13726-13735. DOI: 10.1021/jacs.8b07575
- [3] Inagaki, K. Sugikubo, Y. Miyashita, and C. Mukai, Angew. Chem. Int. Ed. 2010, 49, 2206-2210. DOI: 10.1002/anie.200906994
- [4] Mei, X. Liu, C. Qiao, W. Chen, and C.-C. Li, Angew. Chem. Int. Ed. 2015, 54, 1754-1758. DOI: 10.1002/anie.201410806
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

















No comments yet.