
本文作者:芃洋雪
摘要
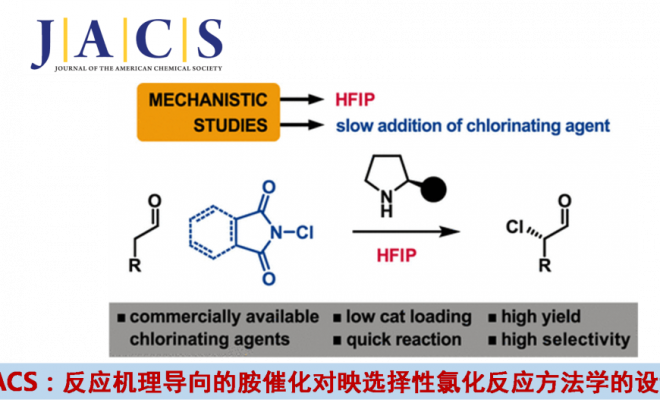
醛的对映选择性氨基催化α-氯化 (enantioselective aminocatalytic α-chlorination)反应方法学,由于无法选择性地产生反应过程中所需的中性中间体,因而,存在较大的挑战。近期,英国Manchester大学 (University of Manchester)的J. Burés课题组在JACS中发表论文,报道采用六氟异丙醇 (HFIP, hexafluoroisopropanol)作为反应溶剂,进而高选择性地形成带电荷中间体 (charged intermediate),从而使原有反应机理发生改变,因而能够采用更为廉价的氨基催化剂 (aminocatalyst)与氯化试剂,对映选择性地完成醛的α-氯化反应,并使反应收率获得进一步提升。同时,催化剂用量减少80-95%,并且,反应条件温和,反应时间显著缩短。
Mechanistically Guided Design of an Efficient and Enantioselective Aminocatalytic α-Chlorination of Aldehydes
G. Hutchinson, C. Alamillo-Ferrer, J. Burés, J. Am. Chem. Soc. 2021, 143, 6805. doi: 10.1021/jacs.1c02997.
正文
在过去20年中,研究人员已经开发出多种不同类型的对映选择性氨基催化反应 (enantioselective aminocatalytic reaction)方法学,进而完成一系列羰基化合物的官能团化反应[1]–[3]。上述方法学实验操作简便,然而,由于相关报道中,更多地关注于发展不同类型的合成转化方法学,而未涉及相同类型反应的进一步优化。因此,当前氨基催化反应方法学的文献报道中,通常需要选择非常规的试剂、较高的催化剂负载、较低的反应温度以及较长的反应时间。例如,醛的不对称α-氯化反应通常需要选择非理想的反应条件,进而确保反应过程具有良好的催化剂转化频率 (TOF, turnover frequency)与良好的立体选择性[4]–[7]。
图片来源:J. Am. Chem. Soc
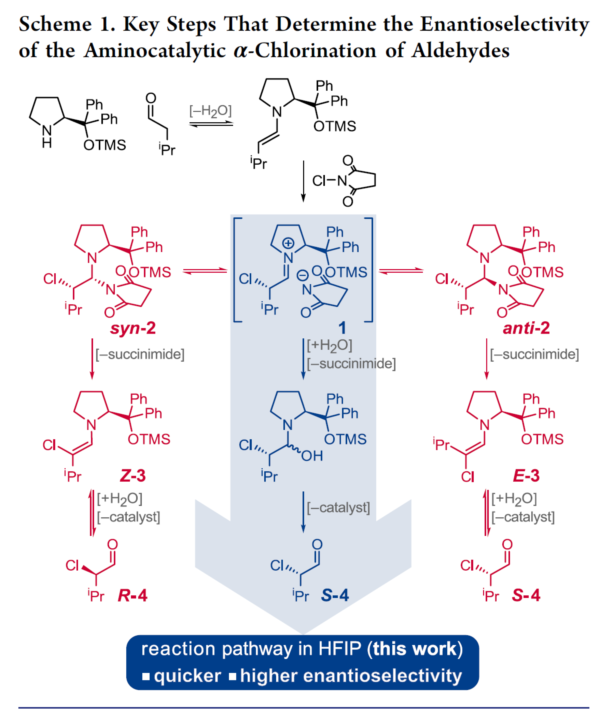
氨基催化反应的相关机理研究已经证实,在反应过程中存在一种非预期的,并涉及中性非对映异位中间体 (neutral diastereotopic intermediate)的反应途径。同时,α-氯化反应的机理研究表明,尽管在氯化步骤中,手性烯胺能够产生优良的对映面选择性控制,然而,由于反应机理路径中出现的的后续分叉 (posterior bifurcation),致使在反应结束后,无法获得优良的对映选择性 (Scheme 1)。其中,催化中间体亚胺盐 (iminium salt) 1能够与1,2-缩醛胺加成物 (1,2-aminal adduct)的非对映异构体syn–2与anti–2之间建立起快速的平衡过程[4]。在标准反应条件下,1,2-缩醛胺加成物由于与带正电荷的亚胺盐1相比,具有更高的稳定性,由此成为反应过程中的静止态 (resting state)。因此,氯化过程通过涉及1,2-缩醛胺加成物的反应途径 (Scheme 1中红色标记的反应途径)进行。并且,与亚胺离子 (iminium ion)直接水解的反应途径 (Scheme 1中蓝色标记的反应途径)相比,速率更为缓慢。此外,1,2-缩醛胺相应非对映异构体的立体专一性消除过程,则分别能够产生烯胺Z-3与E-3,并最终获得在羰基α-位置具有相反绝对构型的产物R4与S4。相关的对映选择性氯化方法学的开创性研究,则通过选择能够有利于形成1,2-缩醛胺中其中一种非对映体的手性胺催化剂[5],并采用能够产生弱配位反荷离子 (poorly coordinating counterions)的氯化试剂[6], [7]以及选用SOMO催化剂[8],或选择具有极高立体位阻的催化剂的组合体系[9],例如N-氯-4-硝基酞酰亚胺(N-chloro-4-nitrophthalimide)与TFA以及乙酸的组合体系,进而有效地解决对映选择性较低的问题。然而,上述各类能够产生对映富集产物的实验方案中,通常需要选择昂贵的试剂或非商业易得的氨基催化剂与氯化试剂,并采用较高的催化剂负载量,有时需要低温反应 (-30 °C),并需要极长的反应时间 (48 h,详见Supporting Information)。
为解决上述实验方案中存在的局限性,近期,英国Manchester大学的J. Burés课题组,采用HFIP (hexafluoroisopropanol),使上述实验方案中相应胺催化中间体 (aminocatalytic intermediate)在有机溶剂中的标准稳定性 (standard stability)发生彻底改变,进而能够有效地完成一系列醛底物的对映选择性α-氯化。

图片来源:J. Am. Chem. Soc
HFIP具有较高的介电常数与较低低的亲核性,能够使阳离子获得稳定化[10]。由此,作者设想与中性的1,2-缩醛胺中间体相比,HFIP更加能够使亚胺阳离子稳定化。为证实上述设想,作者将苯丙醛与Jørgensen-Hayashi催化剂 3a在HFIP溶剂中混合 (Scheme 2)。通过1H-NMR谱学研究,作者观察到Jørgensen-Hayashi催化剂能够定量地转化为苯丙醛亚胺阳离子。而在其它溶剂中,则形成相应的烯胺,例如CD2Cl2, CDCl3, CD3CN, THF-d8, MTBE, toluene-d8, DMSO-d6, CD3OD以及异丙醇 (详见Supporting Information)。
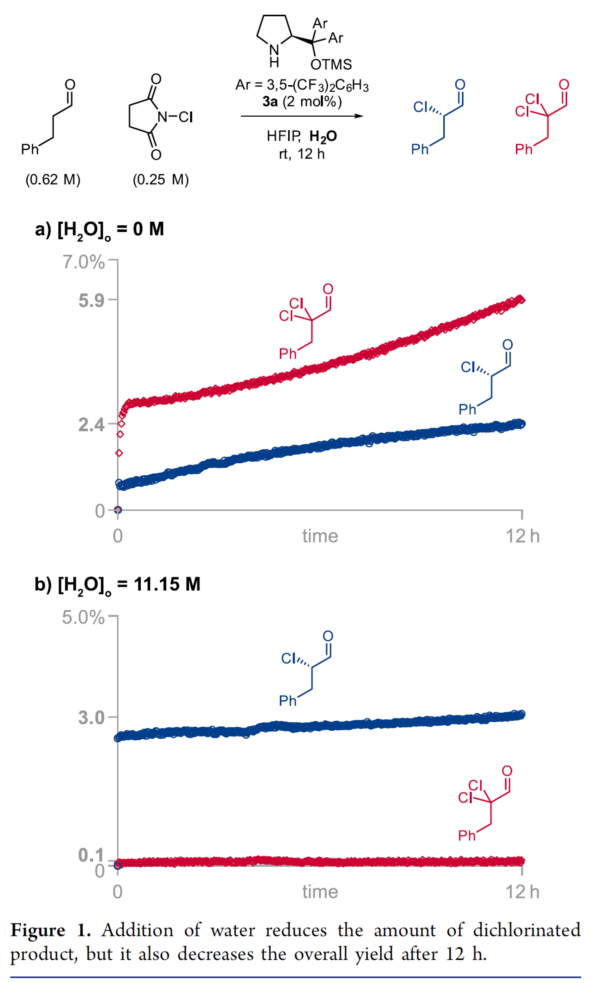
受到HFIP能够使亚胺阳离子稳定化的启发,作者尝试在HFIP溶剂中进行苯丙醛α-氯化反应的研究。最初,作者发现,在NCS以及2.5 eq.苯丙醛与2 mol % Jørgensen-Hayashi催化剂3a作用下,仅能够获得2.4%的单氯化产物与5.9%的二氯化产物 (Figure 1),1H-NMR谱学研究发现,二氯化产物的产生并非与单氯化产物的浓度呈正比 (Figure 1a),这表明二氯化产物主要由催化中间体的过度氯化(overchlorination)产生,并非通过单氯化产物的进一步氯化而产生。同时,作者推测,在加入水之后,能够减少二氯化产物的百分含量,这可能源于水能够参与氯化的亚胺阳离子 (chlorinated iminium)的水解,然而,却无法与氯化的烯胺以及后续的二氯化反应处于平衡状态。接下来,作者进一步观察到,反应在11.15 M的水存在时,二氯化产物的百分含量能够减少至3%,然而,却同时使反应的总产率降低至3% (Figure 1b)。这种产率的降低源自于游离催化剂 (free catalyst)的失活过程,这里作者已经观察到水的加入能够使形成亚胺阳离子的平衡偏向于游离的催化剂 (详见Supporting Information)。
图片来源:J. Am. Chem. Soc
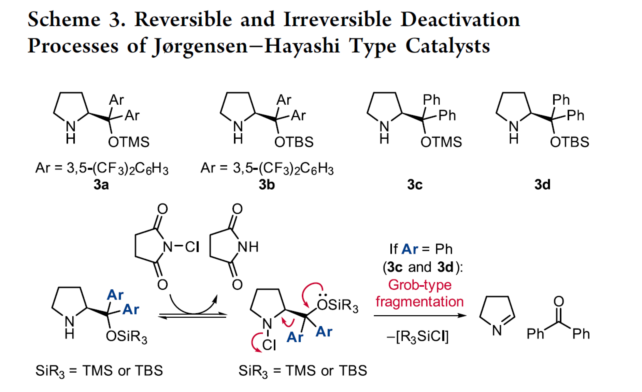
为更好地理解催化剂的失活过程,作者进一步观察反应过程中催化活性中间体1H-NMR信号的变化。在1H-NMR谱图中未观察到醛的相关信号,由此表明,相应的催化中间体能够迅速并近乎定量的形成。接下来,作者通过游离催化剂与氯化试剂之间的反应过程,进而对催化剂失活的机理进行研究。作者通过1H-NMR 波谱对四种不同结构的Jørgensen-Hayashi 催化剂 (3a–3d, Scheme 3) 在HFIP溶剂中与NCS的反应进行监控。实验观察到,带有双重苯基取代的Jørgensen-Hayashi 催化剂对应的氯化中间体能够进一步发生Grob碎片化 (催化剂3c与3d,Scheme 3),而催化剂3a与3b则由于3,5-双(三氟甲基)苯基是存在,不易形成发生Grob碎片化所需的立体电子效应有利的构象,因此,催化剂3a与3b能够稳定存在超过16h。
图片来源:J. Am. Chem. Soc
由于Jørgensen-Hayashi催化剂参与的氯化反应为可逆过程,因此,作者尝试通过加入琥珀酰亚胺,使氯化过程的反应平衡向产生活性催化剂的方向移动,然而,作者发现,加入琥珀酰亚胺的饱和HFIP溶液,并未使反应速率显著提高。最后,作者进一步尝试缓慢加入NCS,从而减少催化剂的失活[11]。根据之前对反应过程中HFIP作用的理解,作者推测,低浓度的NCS不利于失活的氯化催化剂的形成,并能够降低二氯化产物的百分含量。为监控缓慢加入氯化试剂时的反应过程,作者采用原位FT-IR探针 (in situ FT-IR probe),连续测定NCS与琥珀酰亚胺的浓度。研究发现,将NCS加入至含有2.7M水的反应体系的瞬间 (Figure 2a),能够观察到催化剂发生迅速的氯化,并产生9 %收率的目标产物与14%的二氯化产物。延长加入时间至4min (Figure 2b),能够观察到NCS的逐渐累积,同时,目标产物的收率增加至52%,二氯化产物的百分含量降低至2%。而在NCS加入足够缓慢时 (19min, Figure 2c),能够使二氯化产物的百分含量进一步降低至1%。综上所述,通过调整NCS的加入时间,能够进一步提高单氯化产物的收率 (由9%提升至100%),并有效降低二氯化产物的百分含量 (由14%降低至1%)。
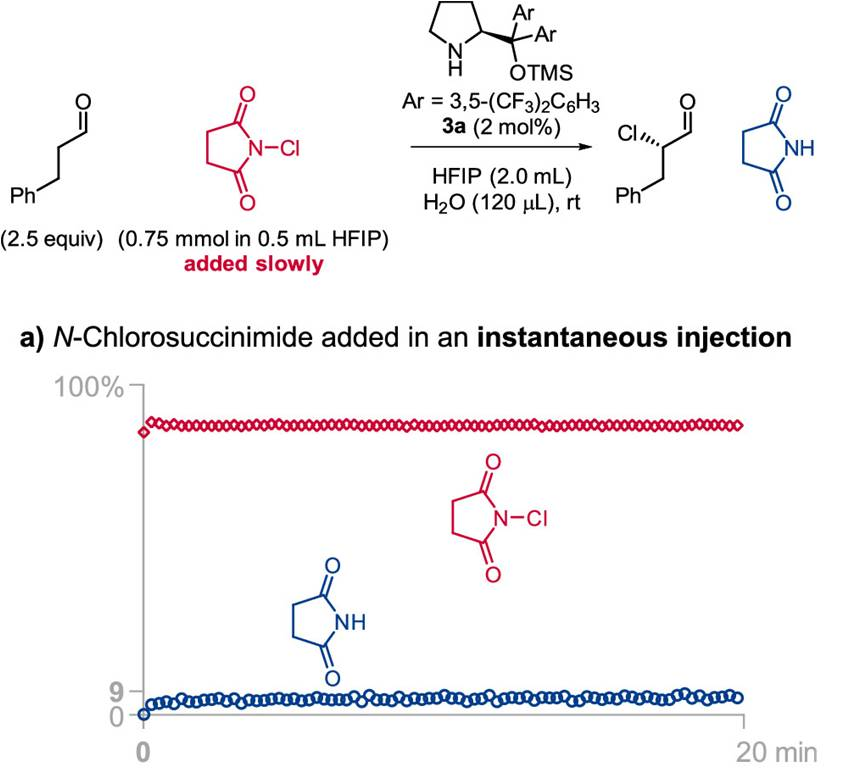
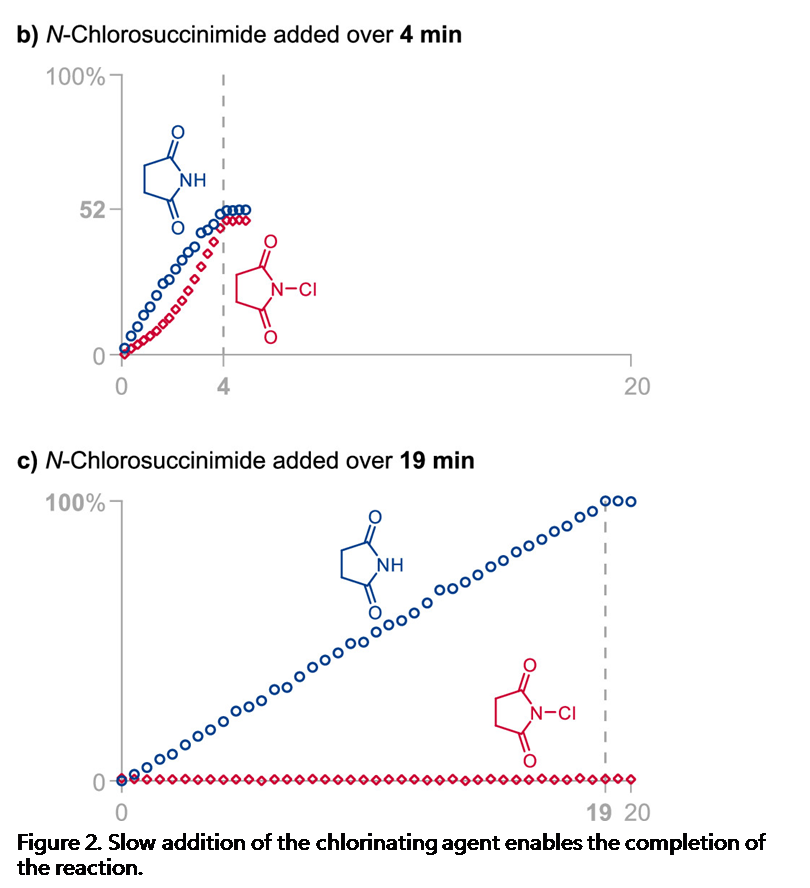
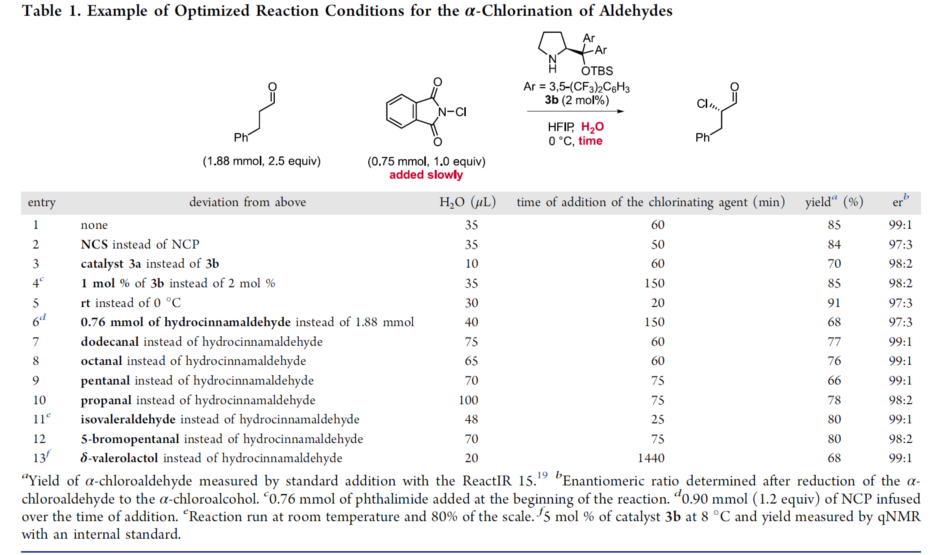
通过上述研究,作者进一步发现,水的浓度与氯化试剂的加入速率为控制反应过程总产率、产物对映体比率以及单氯化与二氯化产物比率的关键参数(详见Supporting Information)。反应体系中水的浓度较高时,能够减少二氯化产物的百分含量,并加速催化剂的氯化过程。并且,较为缓慢地加入氯化试剂,能够减少二氯化产物的百分含量与催化剂的氯化速率。然而,过慢地加入氯化试剂,能够使产物出现外消旋化。同时,作者同样观察到二氯化产物百分含量的增加通常与产物对映比的略微增加存在相关性,可能源自于反应过程中存在的动力学拆分途径 (与醛的α,α-二氟化反应类似) [12]。基于对上述反应机理的理解,作者开始对相关的反应条件进行优化筛选 (Table 1)。研究发现,对于苯丙醛底物,最佳的反应条件为:采用2 mol% 3b作为催化剂,N-氯代邻苯二甲酰亚胺 (NCP, N-chlorophthalimide)作为氯化试剂,反应时间为 60 min, 反应温度为 0 °C (Table 1, entry 1)。之后,作者发现采用NCS作为氯化试剂同样能够获得良好的收率与优良的对映选择性 (Table 1, entry 2)。接下来,作者发现,采用较低选择性的催化剂3a,同样能够获得优良的对映选择性,然而反应收率却有所降低 (Table 1, entry 3)。同时,作者进一步将催化剂3b的用量减少至1 mol%,并延长氯化试剂的加入时间,同时,在反应开始时加入酞酰亚胺,进而减少催化剂的氯化 (Table 1, entry 4)。接下来,该小组发现,将反应温度升至室温,同样能够获得优良的收率与优良的对映选择性 (Table 1, entry 5)。而且,作者进一步将苯丙醛作为限制试剂 (limiting reagent),并将氯化试剂的加入时间调整至超过150 min (Table 1, entry 6),发现同样能够取得优良的对映选择性。这里,延长氯化试剂的加入时间,主要作用是避免催化剂的失活。最后,作者通过进一步调节水的浓度与氯化试剂的加入时间,将上述对映选择性氯化反应方法学进一步应用于月桂醛 (Table 1, entry 7), 辛醛 (Table 1, entry 8), 戊醛 (Table 1, entry 9), 丙醛 (Table 1, entry 10), 异戊醛 (Table 1, entry 11), 5-溴戊醛 (Table 1, entry 12), 以及δ-valerolactol (entry 13),并取得优良的收率与优良的对映选择性。
图片来源:J. Am. Chem. Soc
总结
HFIP作为有机反应的溶剂,能够使带电荷的催化反应中间体稳定化,进而使常规的α-氨基催化氯化 (α-aminocatalytic chlorination reaction)过程的反应机理路径发生改变。通常,这一机理路径的改变,能够有助于获得对映富集的目标产物,然而,却同时伴随二氯化产物的形成与催化剂的失活。通过调节水的浓度与氯化试剂的加入时间,能够有效地解决反应过程中伴随的二氯化产物形成以及催化剂失活的问题。同时,这一全新的醛的α-氯化方法学能够通过选择更为实用的手性催化剂与更加廉价易得的氯化试剂,进而在更加温和的反应条件下,获得优良的收率与优良的对映选择性。
参考文献
(1) B. S. Donslund, T. K. Johansen, P. H. Poulsen, K. S. Halskov, K. A. Jørgensen, Angew. Chem. Int. Ed. 2015, 54, 13860. doi: 10.1002/anie.201503920.
(2) A. V. Penaloza, S. Paria, M. Bonchio, L. Dell´Amico, X. Companyó, ACS Catal. 2019, 9, 6058. doi: 10.1021/acscatal.9b01556.
(3) P. Melchiorre, M. Marigo, A. Carlone, G. Bartoli, Angew. Chem. Int. Ed. 2008, 47, 6138. doi: 10.1002/anie.200705523.
(4) J. Burés, A. Armstrong, D. Blackmond, J. Am. Chem. Soc. 2012, 134, 6741. doi: 10.1021/ja300415t.
(5) N. Halland, A. Braunton, S. Bachmann, M. Marigo, K. A. Jørgensen, J. Am. Chem. Soc. 2004, 126, 4790. doi: 10.1021/ja049231m.
(6) M. P. Brochu, S. P. Brown, D. W. C. MacMillan, J. Am.Chem. Soc. 2004, 126, 4108. doi: 10.1021/ja049562z.
(7) C. Jimeno, L. Cao, P. Renaud, J. Org. Chem. 2016, 81, 1251. doi: 10.1021/acs.joc.5b02543.
(8) M. Amatore, T. D. Beeson, S. P. Brown, D. W. C. MacMillan, Angew. Chem. Int. Ed. 2009, 48, 5121. doi: 10.1002/anie.200901855.
(9) S. Ponath, M. Menger, L. Grothues, M. Weber, D. Lentz, C. Strohmann, M. Christmann, Angew. Chem. Int. Ed. 2018, 57, 11683. doi: 10.1002/anie.201806261.
(10) I. Colomer, A. E. R. Chamberlain, M. B. Haughey, T. J. Donohoe, Nat. Rev. Chem. 2017, 1, 0088. doi: 10.1038/s41570-017-0088.
(11) J. Hein, A. Armstrong, D. G. Blackmond, Org. Lett. 2011, 13, 4300. doi: 10.1021/ol201639z.
(12) J. Franzen, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjærsgaard, K. A. Jørgensen, J. Am. Chem. Soc. 2005, 127, 18296. doi: 10.1021/ja056120u.











No comments yet.