
本文作者:杉杉
导读
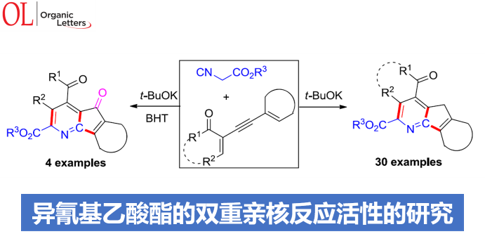
近日,山东师范大学唐波与徐显秀教授课题组在Org. Lett.中发表论文,报道一种碱催化条件下的异氰基乙酸酯与烯炔酮 (enynones)之间的双重环化反应方法学,进而有效地完成一系列4-氮杂芴以及4-氮杂芴酮分子的构建。同时,在这一全新的双重环化策略中,异氰基乙酸酯中的活性亚甲基与异氰基官能团均表现出优良的亲核反应活性。
Dinucleophilic Reactivity of Isocyanoacetate: Base-Catalyzed One-Pot Access to 4-Azafluorenes and 4-Azafluorenones
X. Wang, J. Dong, X. Xu, B. Tang, Org. Lett. 2021 ASAP. doi: 10.1021/acs.orglett.1c03314.
正文
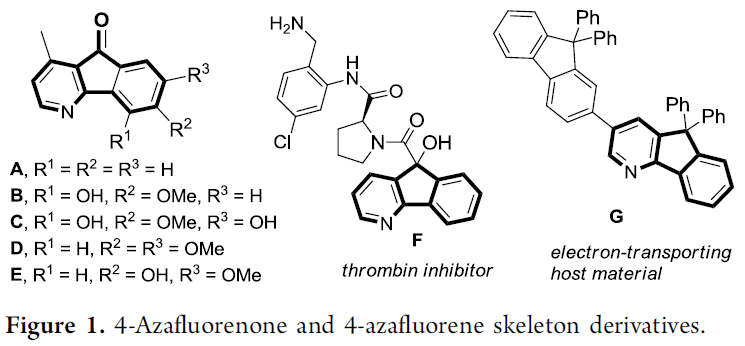
4-氮杂芴与4-氮杂芴酮骨架广泛存在于一系列重要的天然产物、生物活性分子以及合成材料中 (Figure 1)。同时,在构建4-氮杂芴以及4-氮杂芴酮类分子相关的反应方法学研究中,通常采用2-芳基吡啶或芳基(吡啶-3-基)甲酮衍生物进行的关环过程,进而有效地实现上述分子中相应中心五元环的构建[1]。然而,对于构建上述分子的其它反应策略,目前则尚未有相关的文献报道。因此,诸多研究团队开始致力于发展一种全新的、并且更为环境友好的反应策略,进而更加高效地完成上述分子的制备。
其中,具有亲核性C-H结构单元与二价异氰基以及酯基官能团的异氰基乙酸酯分子,目前已经成为杂环分子合成中最为通用的砌块之一。并且,这一分子自1961年发现以来[2],已经成功应用于一系列五元[1]以及六元杂环分子[3]–[6]的构建 (Scheme 1a-1d)。并且,在上述的合成转化过程中,异氰基乙酸酯表现出双重反应活性,即分子中具有酸性的C-H单元作为亲核反应位点,异氰基单元作为亲电反应位点。此外,Zhu课题组研究发现,在异氰基乙酸酯参与的多组分反应中,异氰基与酯基单元均表现出良好的亲核反应活性[7]。
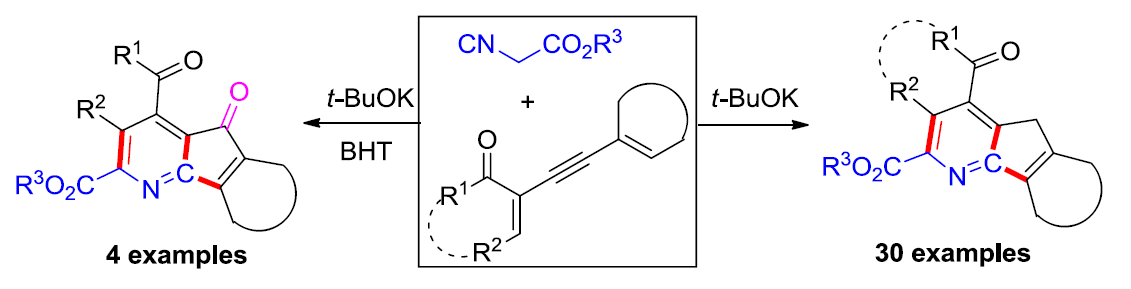
受到上述文献报道以及本课题组前期对于多步domino反应策略中,异氰基乙酸酯分子的亲核 (C-H单元)与亲电 (异氰基单元)反应活性调控的相关研究[8]–[9]的启发。这里,山东师范大学的唐波与徐显秀教授课题组报道一种碱催化条件下的异氰基乙酸酯与烯炔酮之间的双重环化反应方法学,进而成功完成一系列4-氮杂芴以及4-氮杂芴酮衍生物的构建 (Scheme 1e)。值得注意的是,异氰基乙酸酯分子在反应过程中表现出双重的亲核反应活性。同时,环化过程中涉及环腈正离子 (cyclic nitrilium)中间体的形成。

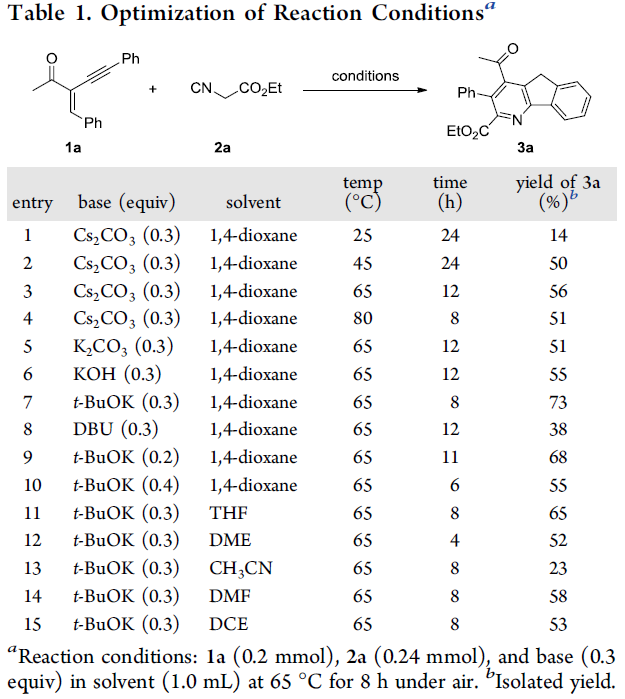
首先,作者采用烯炔酮1a与异氰基乙酸乙酯2a作为模型底物,进行相关环化反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用t-BuOK作为碱,1,4-二噁烷作为反应溶剂,反应温度为65 oC,最终获得73 %收率的目标产物3a。
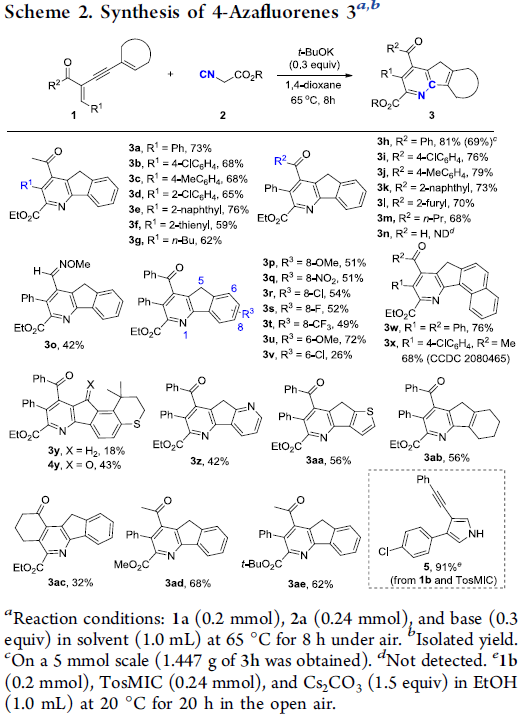
在上述的最佳反应条件下,作者首先对一系列烯炔酮底物应用范围进行考察 (Scheme 2)。研究表明,烯炔酮底物中的R1与R2基团为一系列带有供电子与吸电子基团取代的苯基、萘基以及杂芳基时,均能够顺利参与上述的双重环化过程,并获得相应的4-氮杂芴产物3a–3m (59-81% 收率)。然而,作者发现,上述的标准反应条件对于烯炔醛底物1n,则无法获得预期的双重环化产物3n。而采用相应的烯炔醛肟1o时,却能够获得相应的双重环化产物3o (42% 收率)。同时,该小组进一步发现,上述的标准反应条件对于烯炔酮底物的炔基结构单元中存在的具有一系列供电子与吸电子基团取代的苯基与萘基以及杂芳基与环己烯基,均能够良好地兼容,并获得相应的目标产物3p–3ab (18-76% 收率)。并且,作者观察到,环烯炔酮底物1ac同样能够有效地参与上述的双重环化过程,并以中等程度的反应收率,获得预期的环化产物3ac。
之后,该小组进一步对异氰基乙酸酯底物的应用范围进行深入研究 (Scheme 2)。作者发现,异氰基乙酸甲酯2b与异氰基乙酸叔丁酯2c同样能够有效地完成上述的双重环化过程,并以良好的反应收率获得相应的4-氮杂芴产物3ad与3ae。同时,该小组发现,TosMIC (2d)与烯炔酮1a之间则通过经典的van Leusen吡咯合成反应过程,进而以优良的反应收率,获得相应的3,4-二取代吡咯化合物5,并且,反应过程中并未检测出预期的4-氮杂芴化合物。
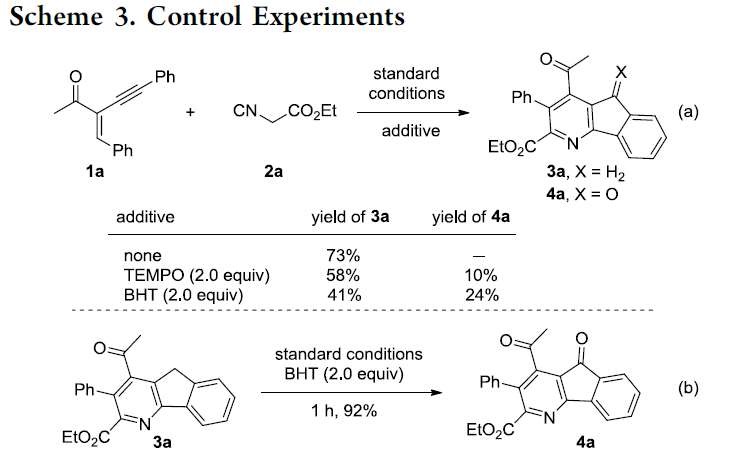
接下来,为提出合理的反应机理,作者进行一系列相关的控制实验研究 (Scheme 3)。首先,该小组发现,向1a与2a的标准反应体系中加入2.0 eq. TEMPO或BHT时,同样能够获得主要的环化产物3a以及少量的产物4a (Scheme 3a)。之后,该小组进一步发现,在产物3a中加入2.0 eq. BHT时,能够顺利转化为产物4a (Scheme 3b)。上述事实表明产物3a的形成过程中并未涉及自由基中间体。同时,反应过程中可能涉及TEMPO或BHT对于3a的氧化。
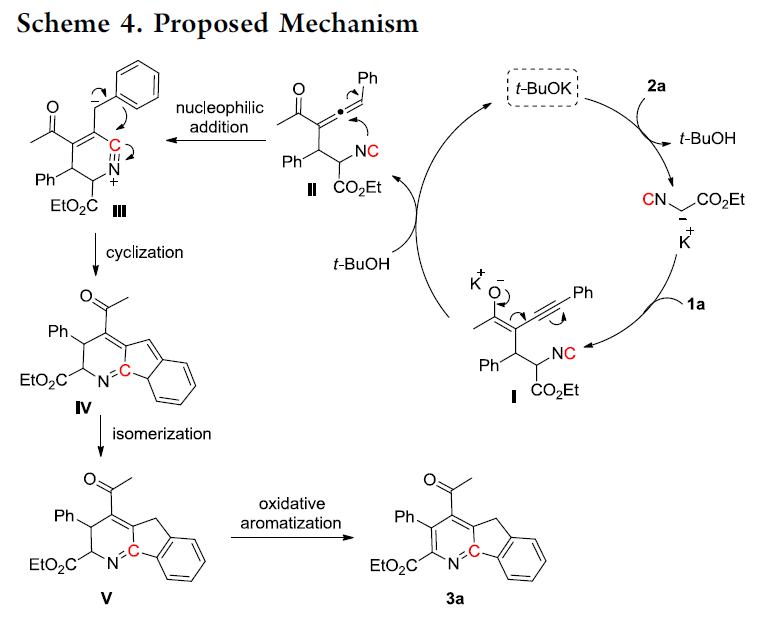
基于上述的控制实验研究以及前期相关的文献报道[10]-[11],作者提出一种合理的反应机理 (Scheme 4)。首先,在t-BuOK存在下,通过异腈2a与烯炔酮1a之间的共轭加成过程,形成负离子中间体I。之后,通过负离子I的质子化过程,形成联烯中间体II。再通过异氰基碳原子与丙二烯中心碳原子之间的分子内亲核加成过程,形成六元环腈正离子中间体III。接下来,中间体III经历进一步的环化过程,形成中间体IV,并通过中间体IV的异构化过程,形成中间体V。最终,通过中间体V进一步的氧化芳构化过程,形成相应的4-氮杂芴酮产物3a。
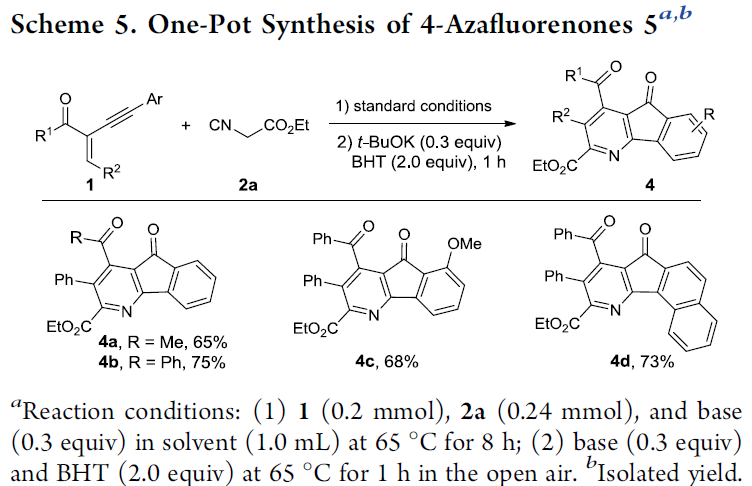
此外,作者进一步发现,在烯炔酮1和异氰乙酸酯2a的标准反应体系中,加入0.3 eq. t-BuOK以及2.0 eq. BHT时,能够以良好至优良的反应收率,获得相应的的4-氮杂芴酮产物4a–4d (Scheme 5)。
总结
山东师范大学唐波与徐显秀教授课题组成功开发出一种全新的异氰基乙酸酯与烯炔酮之间的domino反应方法学,进而成功实现一系列4-氮杂芴与4-氮杂芴酮衍生物的构建。值得注意的是,异氰基乙酸酯分子中的活性亚甲基与异氰基均作为亲核反应位点。同时,机理研究表明,在这一全新的双重亲核加成反应过程中,主要涉及联烯基酮以及六元环腈正离子中间体。并且,这一全新的双重环化策略具有反应条件温和实验操作简洁、原料廉价易得以及高度的官能团兼容性等优势。
参考文献
[1] (a) S. Tu, B. Jiang, H. Jiang, Y. Zhang, R. Jia, J. Zhang, Q. Shao, C. Li, D. Zhou, L. Cao, Tetrahedron 2007, 63, 5406. doi: 10.1016/j.tet.2007.04.053.(b) J. K. Laha, K. P. Jethava, S. Patel, Org. Lett. 2015, 17, 5890. doi: 10.1021/acs.orglett.5b03071.
(c) N. Marquise, V. Dorcet, F. Chevallier, F. Mongin, Org. Biomol. Chem. 2014, 12, 8138. doi: 10.1039/C4OB01629G.
[1] I. Ugi, C. Steinbruckner, IX. Isonitrile, Chem. Ber. 1961, 94, 2802. doi: 10.1002/cber.19610941032. [2] J. Du, X. Xu, Y. Li, L. Pan, Q. Liu, Org. Lett. 2014, 16, 4004. doi: 10.1021/ol501829k. [3] J. Dong, L. Bao, Z. Hu, S. Ma, X. Zhou, M. Hao, N. Li, X. Xu, Org. Lett. 2018, 20, 1244. doi: 10.1021/acs.orglett.8b00186. [4] G. P. Y. Kok, H. Yang, M. W. Wong, Y. Zhao, Org. Lett. 2018, 20, 5112. doi: 10.1021/acs.orglett.8b01948. [5] Z. L. He, C. Wang, Chem. Commun. 2015, 51, 534. doi: 10.1039/C4CC08382B. [6] D. Bonne, M. Dekhane, J. Zhu, Angew. Chem. Int. Ed. 2007, 46, 2485. doi: 10.1002/anie.200605005. [7] (a) W. Li, X. Yu, Z. Yue, J. Zhang, Org. Lett. 2016, 18, 3972. doi: 10.1021/acs.orglett.6b01737.(b) J. Li, Y. Liu, C. Li, X. S. Jia, Chem. – Eur. J. 2011, 17, 7409. doi: 10.1002/chem.201100977.
(c) M. Charaschanya, K. Li, H. F. Motiwala, J. Aubé, Org. Lett. 2018, 20, 6354. doi: 10.1021/acs.orglett.8b02531.
[8] (a) J. Tan, X. Xu, L. Zhang, Y. Li, Q. Liu, Angew. Chem. Int. Ed. 2009, 48, 2868. doi: 10.1002/anie.200805703.(b) Y. Li, X. Xu, J. Tan, C. Xia, D. Zhang, Q. Liu, J. Am. Chem. Soc. 2011, 133, 1775. doi: 10.1021/ja110864t.
(c) X. Xu, L. Zhang, X. Liu, L. Pan, Q. Liu, Angew. Chem. Int. Ed. 2013, 52, 9271. doi: 10.1002/anie.201303604.
(d) L. Zhang, X. Xu, J. Tan, L. Pan, W. Xia, Q. Liu, Chem. Commun. 2010, 46, 3357. doi: 10.1039/C001617A.
(e) J. Dong, X. Wang, H. Shi, L. Wang, Z. Hu, Y. Li, X. Xu, Adv. Synth. Catal. 2019, 361, 863. doi: 10.1002/adsc.201801103.
[9] (a) Z. Hu, H. Yuan, Y. Men, Q. Liu, J. Zhang, X. Xu, Angew. Chem. Int. Ed. 2016, 55, 7077. doi: 10.1002/anie.201600257.(b) Z. Hu, J. Dong, Y. Men, Z. Lin, J. Cai, X. Xu, Angew. Chem. Int. Ed. 2017, 56, 1805. doi: 10.1002/anie.201600257.
[10] W. Li, X. Yu, Z. Yue, J. Zhang, Org. Lett. 2016, 18, 3972. doi: 10.1021/acs.orglett.6b01737. [11] (a) J. Li, Y. Liu, C. Li, X. Jia, Chem. – Eur. J. 2011, 17, 7409. doi: 10.1002/chem.201100977.(b) M. Charaschanya, K. Li, H. F. Motiwala, J. Aubé, Org. Lett. 2018, 20, 6354. doi: 10.1021/acs.orglett.8b02531.



















No comments yet.