
本文来源于四川大学化学学院和余达刚教授
导读:
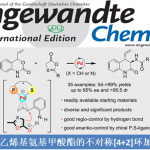
近日,四川大学余达刚教授课题组在《Nature Catalysis》发表了题为“Visible-light photocatalytic di- and hydro-carboxylation of unactivated alkenes with CO2”的文章,四川大学为第一单位,化学学院余达刚教授为通讯作者,2021届博士宋磊和2021级博士生王伟为共同第一作者。特别感谢巴塞尔大学O. S. Wenger, F. Glaser和 B. Pfund以及弗吉尼亚大学的J. J. Chruma教授的宝贵建议。衷心感谢国家自然科学基金委、四川省科技厅、四川大学和北京分子科学中心的经费支持。
Visible-light photocatalytic di- and hydro-carboxylation of unactivated alkenes with CO2
Lei Song, Wei Wang, Jun-Ping Yue, Yuan-Xu Jiang, Ming-Kai Wei, Hai-Peng Zhang, Si-Shun Yan, Li-Li Liao & Da-Gang Yu*
Nature Catalysi, 2022, ASAP.doi: 10.1038/s41929-022-00841-z
正文:
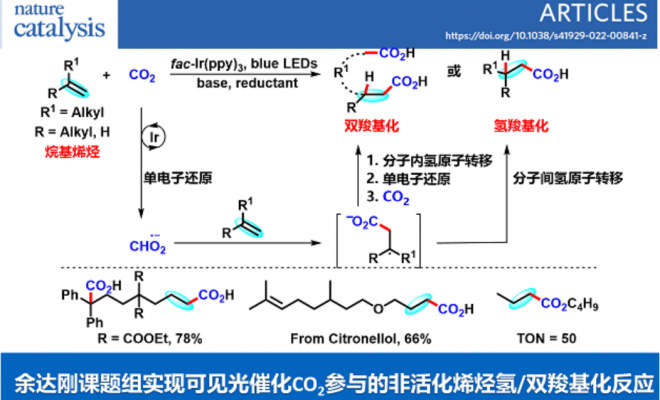
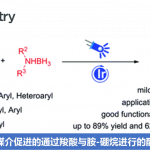
二氧化碳(CO2)是一种对气候变化有重要影响的温室气体,也是一种储量丰富、廉价易得、无毒且可再生的碳一资源。利用CO2参与化学转化,高效规模化制备高附加值化学品和大宗化学品,是服务国家重大战略需求的重要方式,对促进资源开发和可持续发展具有重要意义。其中,利用CO2精准合成在医药和材料等领域有重要应用的羧酸及其衍生物,是合成化学领域的重要研究方向,虽然已有可喜进展,但仍然存在很多问题。另一方面,太阳能是取之不尽用之不竭的清洁能源,可见光催化具有绿色环保、条件温和以及官能团兼容性高等特点,因此备受关注。考虑到烯烃来源广泛,近年来,人们利用可见光催化烯烃与CO2反应,实现了一系列具有不同化学和区域选择性的羧基化反应,合成了多种类型的重要羧酸产物。然而,在已报道的例子中,底物主要局限于活化烯烃(例如芳基乙烯和丙烯酸酯)。相比而言,非活化烯烃由于反应性更低,通常比活化烯烃更难与CO2发生羧基化反应。因此,开发一种新策略来实现可见光催化非活化烯烃和CO2的羧基化反应具有重要意义。
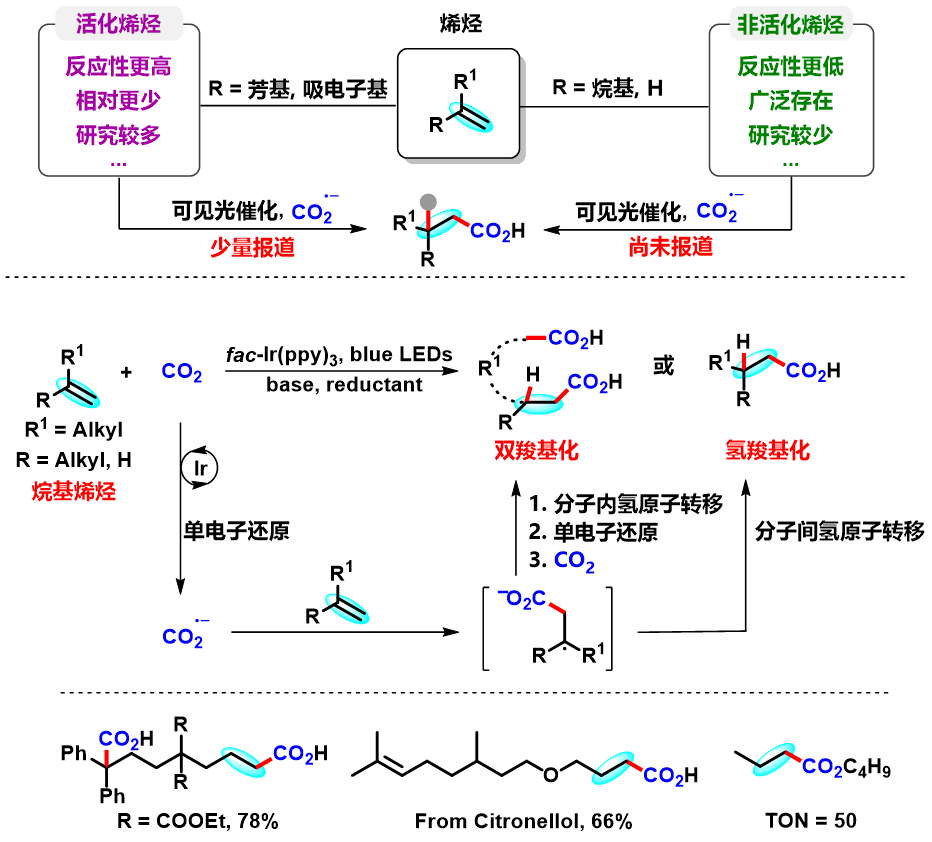
不同于常见的CO2双电子活化方式(亲核试剂直接进攻CO2),CO2单电子活化方式(CO2单电子还原形成CO2自由基负离子)具有独特的反应中间体和反应路径,可以实现不同区域和化学选择性的羧基化反应,从而为重要羧酸化合物合成提供新型合成方法。然而,由于CO2自身还原电势[E1/2 (CO2/CO2•−) = -2.21 V vs SCE]较高,单个可见光光子能量有限,利用常规的可见光催化体系很难将其直接还原,导致目前可见光催化CO2单电子活化的报道较少。此外,在该研究之前,还没有通过可见光催化实现CO2自由基负离子进攻普通烯烃的报道。针对以上问题,四川大学化学学院余达刚课题组在前期可见光促进CO2转化研究(研究总结:Acc. Chem. Res. 2021, 54, 2518)基础上,成功实现了可见光催化CO2参与非活化烯烃的氢羧基化和远程双羧基化。该工作利用连续光致电子转移策略(Consecutive Photo-induced Electron Transfer, ConPET),将CO2还原为CO2自由基负离子,进攻非活化烯烃得到相应的烷基自由基。该烷基自由基可以发生分子间氢原子转移过程,得到选择性氢羧基化产物;在合理设计的底物中,该烷基自由基也可以发生分子内的氢原子转移(HAT),进而发生单电子还原形成碳负离子,进攻另一分子CO2得到相应的二元酸及其衍生物。作者也进一步对该新颖的反应路径进行了详细的实验研究,通过机理研究证实了相关中间体的存在,并从多方面佐证了ConPET机制还原CO2的过程。该反应具有条件温和、底物适用性广和官能团兼容性好等优点。一系列烷基烯烃(包括丙烯和工业来源的混合烯烃)都能较好地参与该反应,以中等至优秀的产率得到一元羧酸、二元羧酸和非天然α-氨基酸衍生物。
总的来说,该研究首次实现了可见光催化CO2参与的非活化烯烃的选择性氢/双羧基化反应。该体系通过连续光致电子转移策略实现了CO2单电子活化,为CO2高值化利用提供了新思路,也为脂肪酸和二酸单体等重要羧酸的合成提供了新途径,有望应用于医药、农药、食品和材料等领域。
四川大学为第一单位,化学学院余达刚教授为通讯作者,2021届博士宋磊和2021级博士生王伟为共同第一作者。
相关连接
余达刚团队实现可见光催化CO2参与的非活化烯烃氢/双羧基化反应
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载














No comments yet.