
本文作者:杉杉
导读
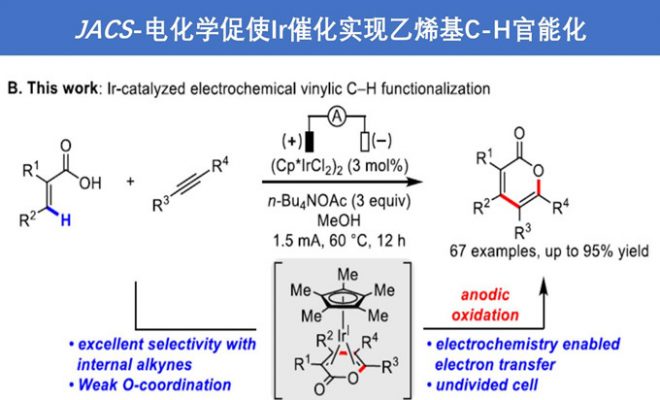
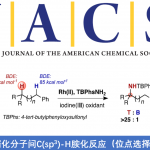
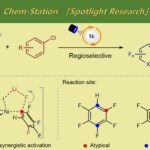
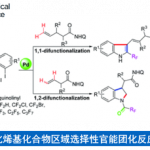
电化学与金属催化协同作用已成为C-H官能化的强大工具,然而该反应主要限于芳烃C-H官能化。近日,上海有机所梅天胜课题组在JACS发表论文,报告Ir催化下,通过电化学乙烯基C-H官能化,实现丙烯酸与炔烃的偶联。在该反应中,铱催化剂可实现C-H/O-H炔烃的环化,获得优异收率的α-吡喃酮。初步机理表明,阳极氧化二烯-铱(I)配合物(配位饱和的18电子配合物)对于释放产物和再循环Ir(III)中间体至关重要。重要的是,反应条件一致(不存在电流),常见的化学氧化剂(如Ag(I)或Cu(II))不会获得相应的产物。
Electrochemistry-Enabled Ir-Catalyzed Vinylic C–H Functionalization
Qi-Liang YangYi-Kang XingXiang-Yang WangHong-Xing MaXin-Jun WengXiang YangHai-Ming GuoTian-Sheng Mei*
JACS, APSP, DOI:10.1021/jacs.9b11915

正文
近年来,电化学有机合成作为一种热门的领域,该方法主要利用电流代替常规的化学氧化剂或还原剂来促进反应,同时可以通过电势的调节,实现独特的反应性和化学选择性。C-H官能化作为电化学反应作为典型的代表,因为这种方法不仅避免了底物的预官能化,而且在逆合成分析中提供了新颖的方法。由于C-H键的氧化电位高于常见官能团和有机溶剂,通常采用介体参与电化学氧化。例如,Baran开发了一种四氯-N-羟基邻苯二甲酰亚胺(Cl4NHPI)作为介体,实现可持续的电化学烯丙基氧化。另外,通过与Pd催化结合可实现选择性C(sp3)-H官能化。同样,一些金属催化(如Pd、Ni、Co、Ru、Cu、Rh、Ir等)促进电化学芳烃C-H官能化也被报道。在这些文献中,电流通常作为氧化剂促进催化反应。
然而,电化学乙烯基C-H官能化文献报道的很少。一般而言,烯烃π-键的电化学氧化要比官能化乙烯基C-H键容易得多,因为π-键的解离能(大约65 kcal/mol)比乙烯基C-H键(大约111 kcal/mol)低(Scheme 1A)。基于上述的总结,上海有机所梅天胜研究员课题组报道了Ir催化下,实现炔烃与丙烯酸的环化反应,获得多种α-吡喃酮衍生物(Scheme 1B),此结构在天然产物和生物活性小分子中普遍从存在。尽管一些金属催化(如Co、Ru、Rh和Pd)实现了丙烯酸与炔烃的C-H环化反应已有报道,但Ir催化尚未被报道,可能是由于脱羧作用或反应过程中形成的稳定的18电子二烯-Ir(I)络合物导致Ir催化剂离解困难。
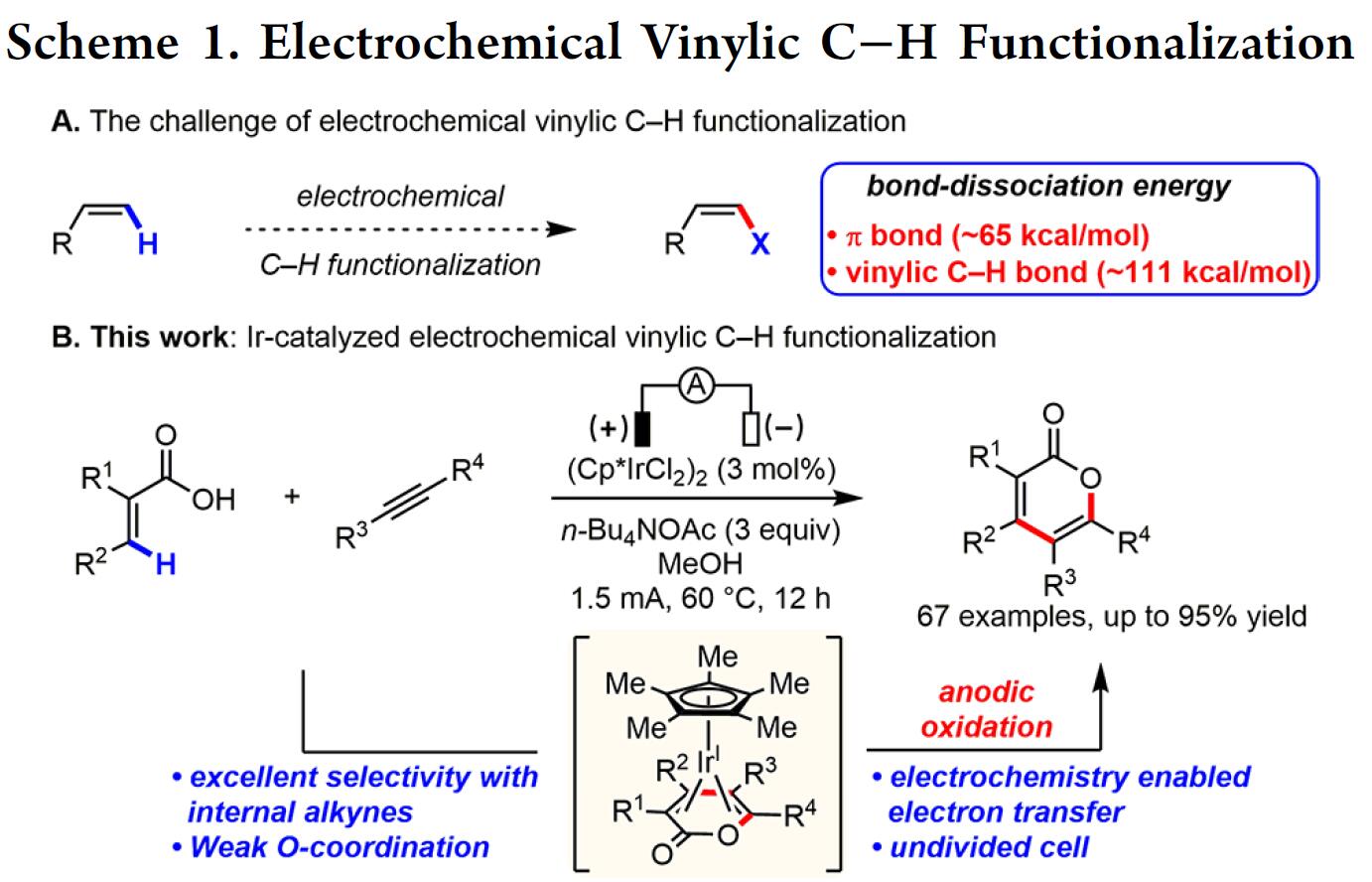
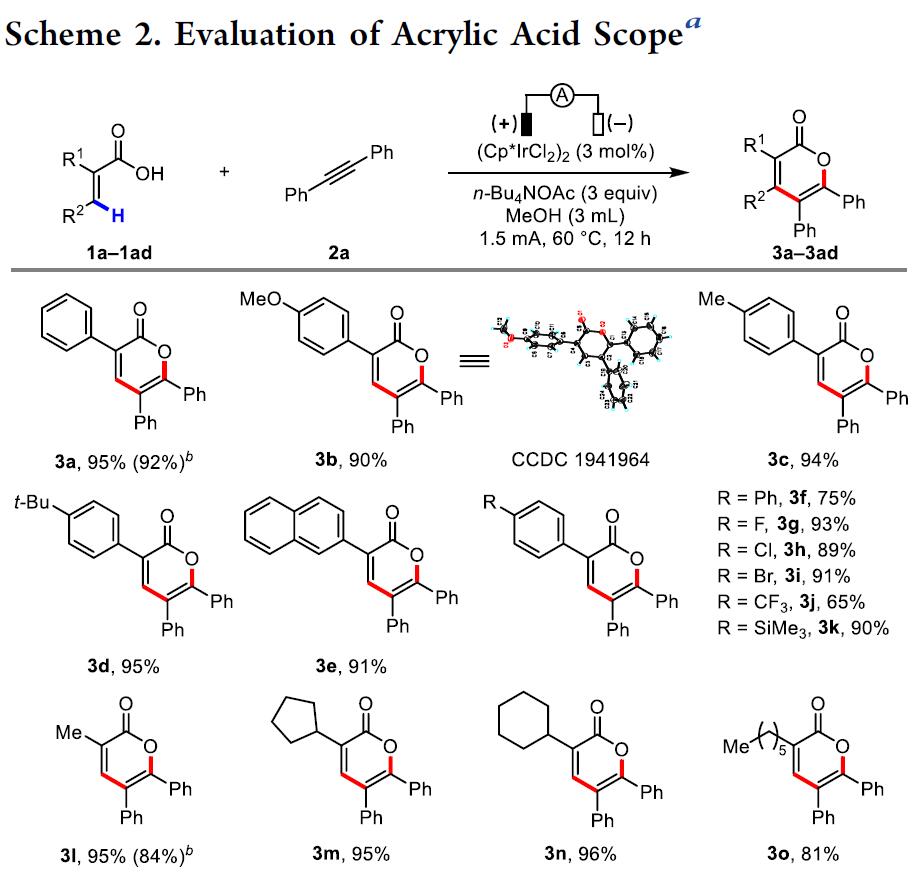
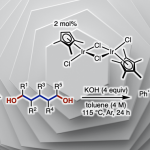
首先,作者以2-苯基丙烯酸(1a)和二苯基乙炔(2a)作为模型底物进行筛选(Table 1)。作者发现,以(Cp*IrCl2)2(3 mol%)为催化剂、n-Bu4NOAc(3eq)作为碱、甲醇作为溶剂,在1.5 mA下恒流电解条件下,60°C反应12h可获得分离产率为95%的吡喃酮3a(entry 1)。当使用其他溶剂时,收率显着降低(entries 2–5),增加电流(entries 6-7)或降低反应温度会导致产率略低(entries 8-9)。令作者高兴的是,在室温下用IKA ElectroSyn 2.0(电化学合成仪)进行反应时,同样可以实现上述反应(entry 10)。而对不同的电解质筛选中,作者发现乙酸根阴离子对反应至关重要(entries 11-13),这些结果表明乙酸盐协助C-H活化中去质子过程。值得注意的是,在其他条件相同的情况下,在没有电流时,化学氧化剂(O2、Ag2CO3和Cu(OAc)2)无法提供所需的产物(entries 14-16),仅有PhI(OAc)2作为氧化剂时,获得3a的产率为18%(entry 17)。最后,控制实验表明,铱催化剂和电流对于该反应都是必不可少的(entries 18-19)。
在获得上述最佳反应条件后,作者开始对底物范围进行了扩展。首先,以二苯基乙炔(2a)与α或β-取代丙烯酸进行反应(Scheme 2)。反应结果表明,α-取代丙烯酸反应不受电子效应的影响(3a–3v),均能获得较高收率的α-吡喃酮衍生物,一些活性的基团(如卤化物、烯丙基、酯基等)也能够兼容,未取代的丙烯酸给出良好的产率(3w)。相反,β-取代的丙烯酸通常给出较低的产率(3x–3ac),在这些情况下,反应12小时后,可以观察到部分未反应的炔烃,但是丙烯酸通常被完全消耗掉。有趣的是,在α/β-位置同时带有甲基时,可获得良好的收率(3ad)。

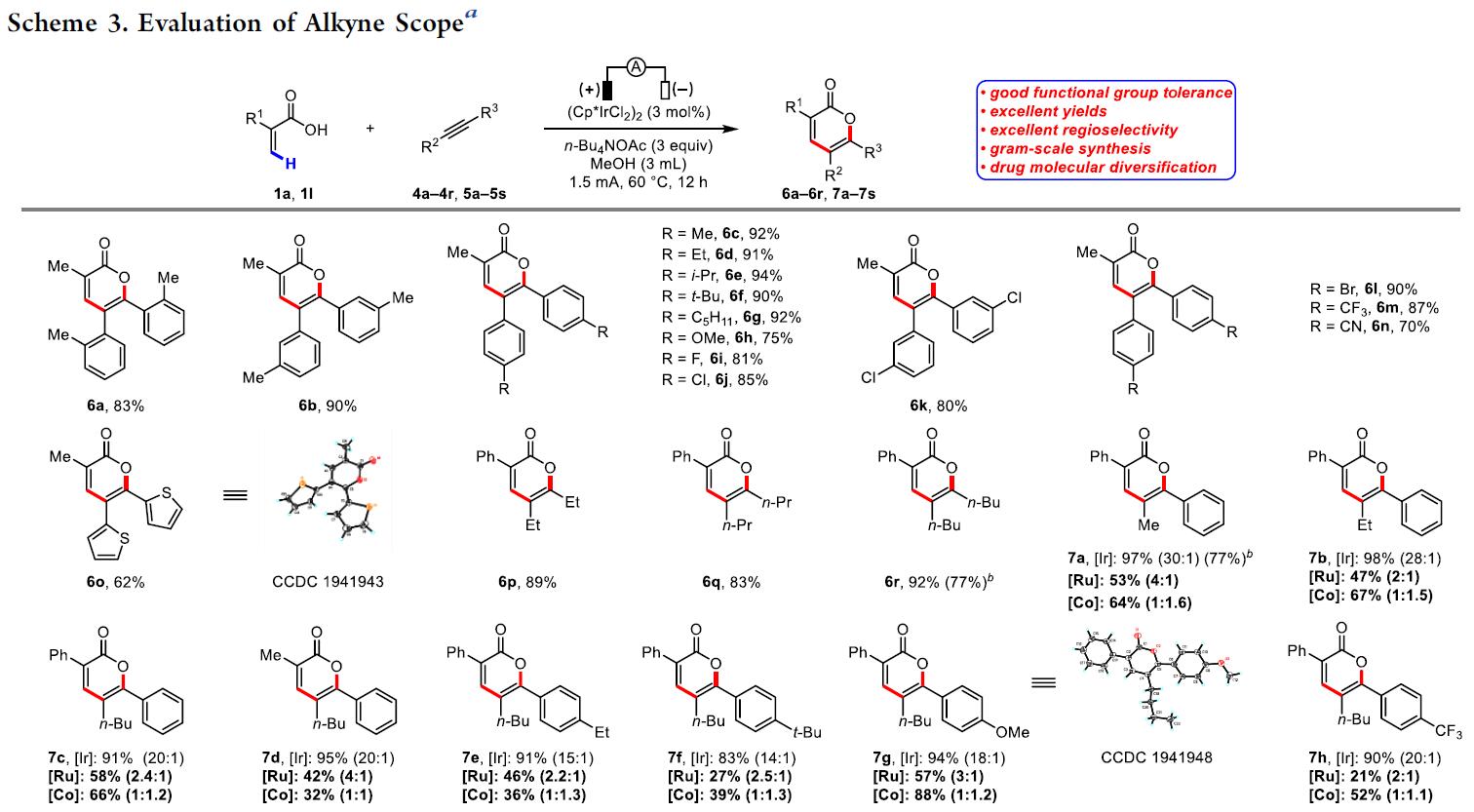
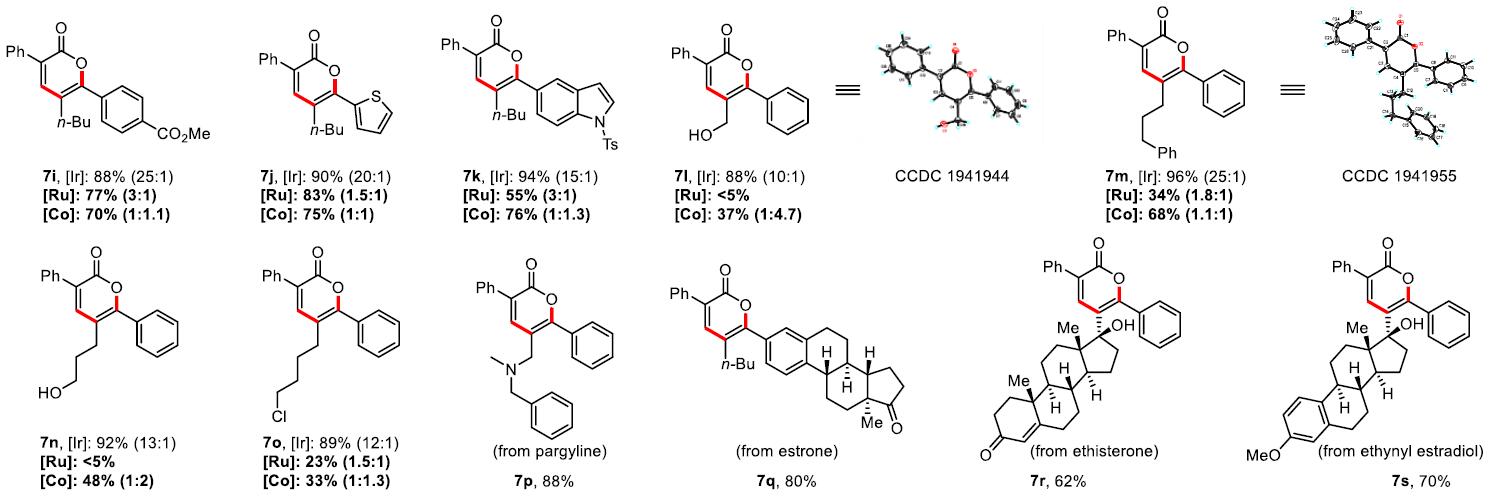
接下来,作者继续扩展内部炔烃底物范围(Scheme 3)。反应结果表明,取代二芳基乙炔(6a–6n)与二烷基乙炔(6o–6q)均可获得较高收率的α-吡喃酮衍生物。令作者高兴的是,使用不对称炔烃则表现较高的区域选择性,反应结果总是将芳烃部分置于氧杂原子附近(7a–7o)。与已知具有不对称内部炔烃的Co或Ru催化的丙烯酸环化反应相比,一般来说,钴催化的化学氧化剂环化反应具有1:1-1:5的区域选择性,钌催化的化学氧化剂环化反应具有1:1-4:1的区域选择性,相比之下,上述方案可产生10:1-30:1的区域选择性。这清楚地证明了该反应可通过不对称内部炔烃实现高度选择性的区域选择性。此外,该反应同样可对天然产物进行后期修饰(7p–7s)。
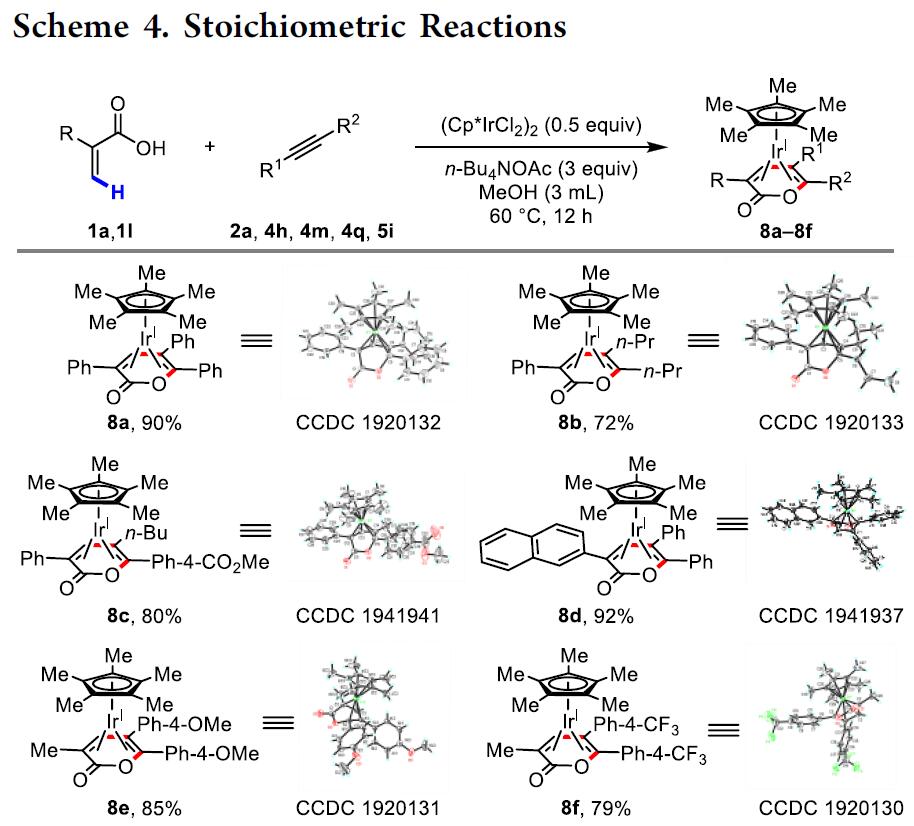
为了进一步了解电化学乙烯基C-H环化反应机理,作者做了一些对照实验(Scheme 4)。首先,作者研究了在没有电流的情况下丙烯酸、炔烃和(IrCp*Cl2)2的反应,有趣的是,该条件下可获铱夹层的复合物8a-8f(配位饱和的18电子配合物),这说明了它们对化学氧化剂的稳定性和惰性。
随后,通过使IrCp*(OAc)2(DMSO)络合物9与底物1a在60℃下反应2h合成络合物10,再与炔2a反应得到配合物8a(Scheme 5A),铱配合物8a作为电化学乙烯C-H环化反应的有效中间体和催化剂(Scheme 5B)。有趣的是,在60℃或120℃下用代表性的化学氧化剂Ag2CO3和Cu(OAc)2与8a反应获得极低收率的3a,但当通入电流后产率提高至84%(Scheme 5C)。

基于上述的实验以及文献的总结,作者提出了一种可能的反应机理(Scheme 6)。首先,中间体A与1a经C-H活化得到Ir(III)中间体B,经配体交换获得配合物C,随后炔烃的插入形成七元铱配合物D,经过还原消除得到Ir(I)配合物E。中间体E是配位饱和的18电子配合物,在阳极氧化时,从E得到产物3a,并且使配合物A循环。

总结
上海有机所梅天胜课题组首次发现Ir催化下,通过电化学乙烯基C-H官能化,实现丙烯酸与炔烃的偶联。在该反应中,铱催化剂可实现C-H/O-H炔烃的环化,获得优异收率的α-吡喃酮。初步机理表明,阳极氧化二烯-铱(I)配合物(配位饱和的18电子配合物)对于释放产物和再循环Ir(III)中间体至关重要。此外,与其他文献相比,该反应具有高度的区域选择性。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

















No comments yet.