
本文作者:杉杉
导读
α-手性炔基化合物是构成诸多生物活性化合物、化学探针以及功能材料的关键结构单元,并且,该化合物同样能够作为有机合成中的重要合成子。这里,南京大学朱少林课题组报道一种通过NiH催化的烯基化合物与溴代炔烃之间的还原迁移氢炔基化 (reductive migratory hydroalkynylation)反应方法学,并以较高的反应收率与优良的区域选择性,获得相应的苄位炔基化产物。同时,作者发现,在采用手性PyrOx配体时,能够进一步实现各类苯乙烯底物的催化对映选择性氢炔基化反应,进而获得对映富集的苄位炔基化合物。并且,相应的手性苄位炔基化合物能够进一步转化为一系列不对称合成中较为关键的手性合成子。
Nickel-catalysed migratory hydroalkynylation and enantioselective hydroalkynylation of olefins with bromoalkynes
X.Jiang, B.Han, Y. Xue, M. Duan, Z. Gui, Y. Wang, S. Zhu, Nature Commun.2021, 12, 3792. doi: 10.1038/s41467-021-24094-9.
正文
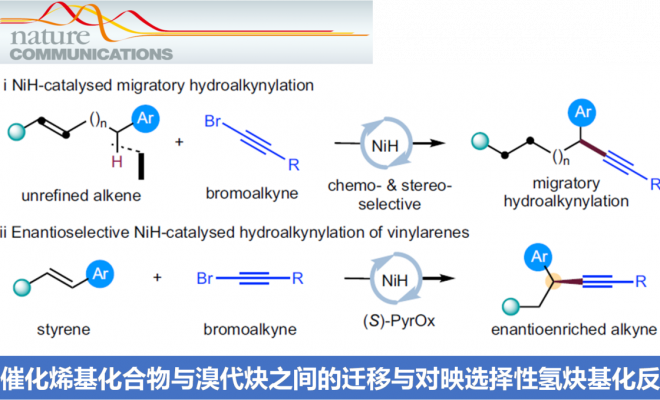
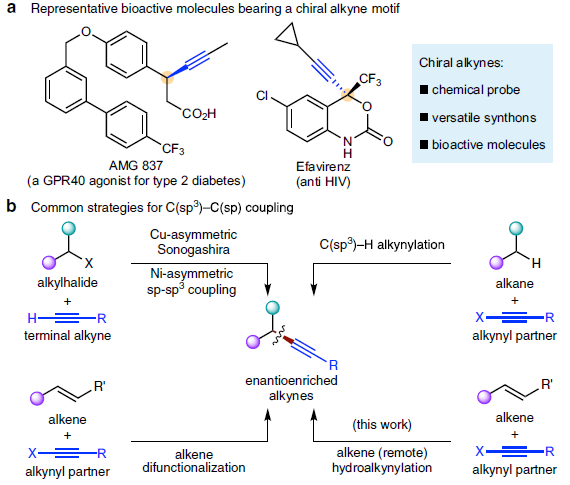
α-手性炔基化合物作为重要的结构单元,广泛存在于诸多生物活性化合物、化学探针以及功能材料分子中 (Fig. 1a)。同时,作为有机合成中重要的合成子,α-手性炔基化合物能够实现多种不同类型的合成转化过程。长期以来,催化对映选择性C(sp3)-C(sp)偶联方法学,作为构建α-手性炔基化合物的有效策略而备受关注 (Fig. 1b)。例如,Liu课题组[1]-[2]报道Cu催化的不对称Sonogashira C(sp3)-C(sp)偶联反应策略。Shi[3]与Liu课题组[4]分别报道采用Pd与Cu催化剂促进的不对称催化C(sp3)-H炔基化反应方法学。Liu课题组[5]报道通过铜催化的烯键双官能化策略,进而构建一系列对映体富集的炔基化产物。Suginome (杉野目 道紀 )课题组[6]报道首例镍催化的1,3-二烯底物的不对称氢炔基化反应方法学。近年来镍催化剂由于具有低成本、易于进行氧化加成以及氧化态多样性等优势,作为钯催化剂的有力补充,已经广泛应用于涉及C(sp3)结构片段的交叉偶联反应方法学研究。近期,采用氢化镍[7]-[9]催化的还原迁移氢官能团化反应方法学 (reductive migratory hydrofunctionalization) [10]-[13]已经发展成为实现远程C(sp3)-H键选择性官能团化的有效策略。与传统的交叉偶联策略相比,上述策略中采用廉价易得,并具有较高稳定性的烯基化合物或烯基化合物前体作为起始原料,代替需要特殊方法制备的金属有机试剂,并能够有效地实现ipso-位置以及远程C(sp3)-H位点的选择性官能团化。在此,本文报道一种采用NiH催化烯基化合物与溴代炔之间的还原迁移氢炔基化反应方法学 (Fig. 1c, i)。同时,作者发现,在采用手性PyrOx配体时,能够进一步实现苯乙烯底物的催化对映选择性氢炔基化反应,并获得一系列对映富集的苄位炔基化合物 (Fig. 1c, ii)。

![]()
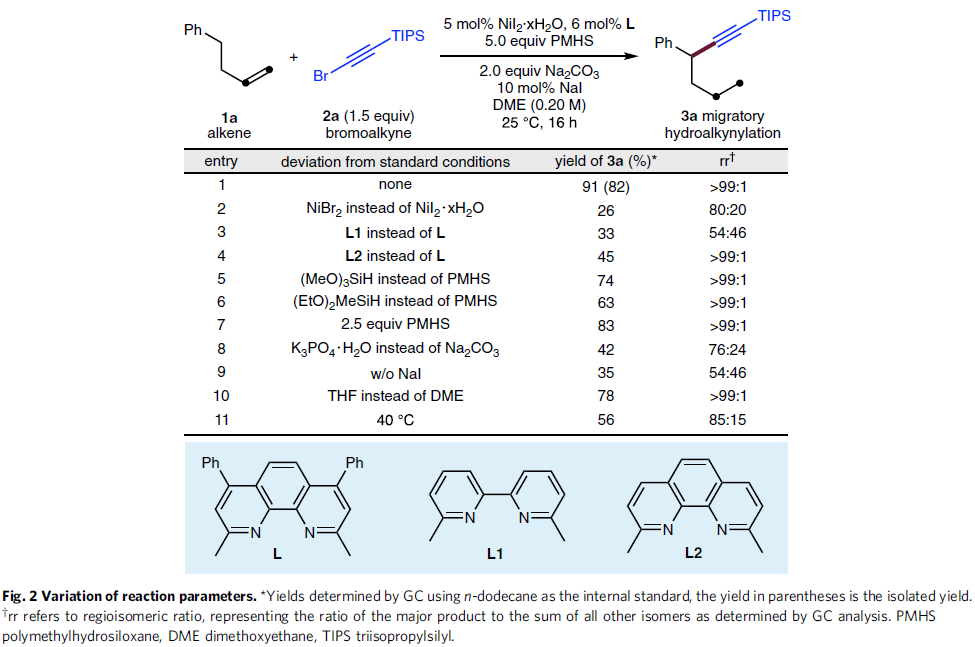
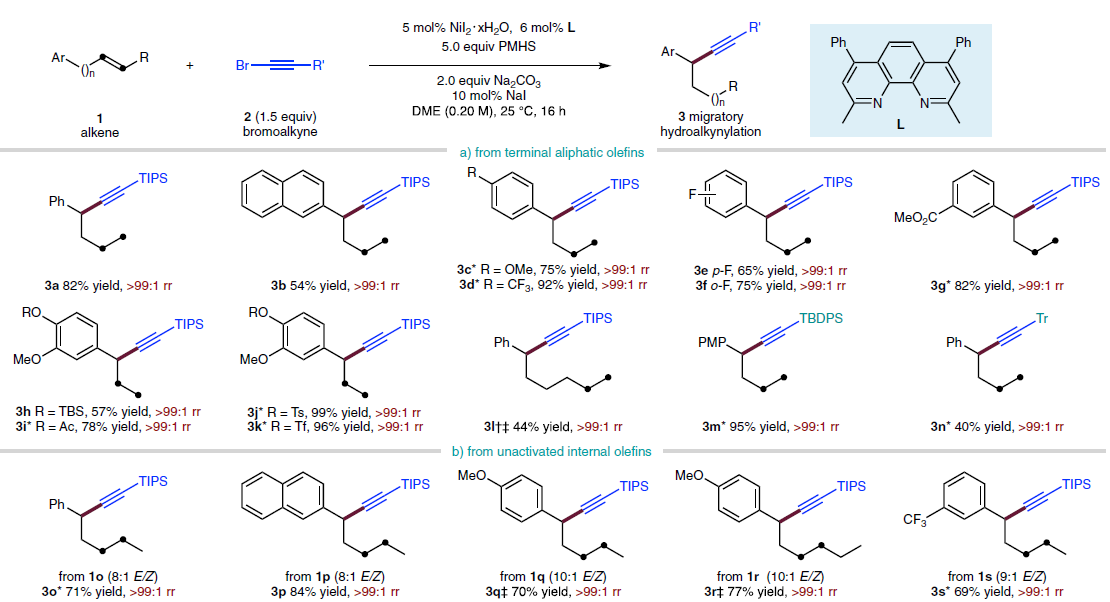
首先,作者采用4-苯基-1-丁烯1a与炔基化试剂2a作为模型底物,进行氢炔基化反应条件的优化筛选 (Fig. 2)。进而确定最佳的反应条件为:采用5 mol% NiI2·xH2O作为催化剂,6 mol% L作为配体,2eq. Na2CO3作为碱,5eq. PMHS作为还原剂,10 mol% NaI作为添加剂,在DME溶剂中25 ℃下进行反应,最终,以91%反应收率与>99:1 rr (regioisomeric ratio)获得相应炔基化产物3a。
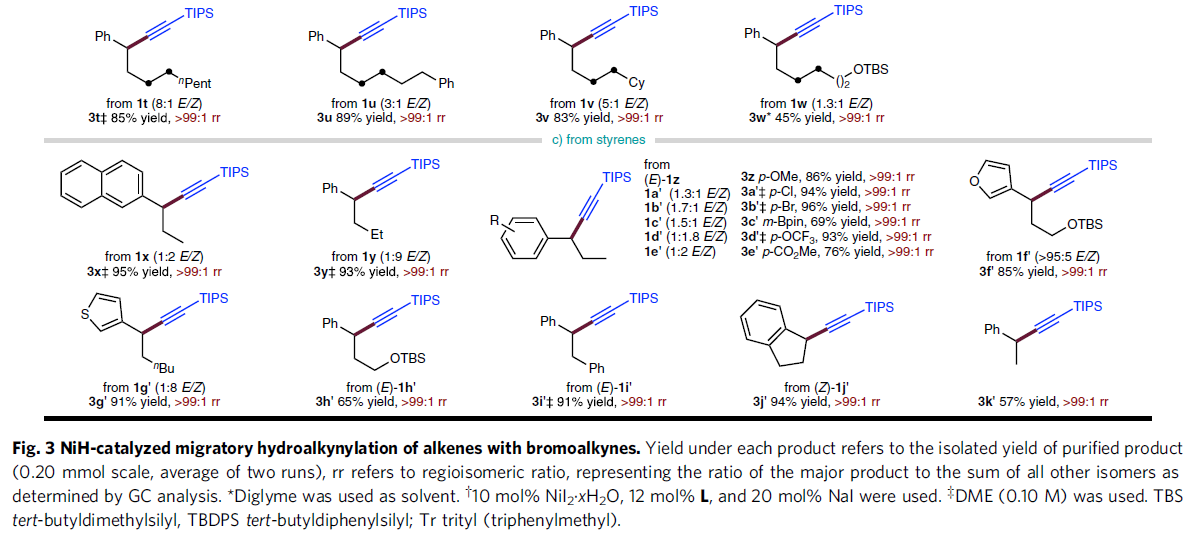
在获得上述最佳反应条件后,作者对烯基化合物1的底物应用范围进行研究 (Fig. 3)。研究发现,远端芳环 (remote aryl ring)中有供电子 (3c)或吸电子 (3d–3g)基团取代的非活化端烯 (unactivated terminal alkene)底物,均能够顺利地参与上述反应过程。同时,上述的最佳反应条件对于芳环远端位置带有醚基团 (3c,3h–3k,3m)、三氟甲基 (3d)以及酯基团 (3g,3i)取代的非活化端烯底物,同样能够良好地兼容。并且,作者发现,在C=C键与远端芳基之间的链长增加 (3l)之后,同样能够获得相应的苄位氢炔基化产物,然而,反应收率却显著降低。值得注意的是,上述的标准反应条件对于甲硅烷基以及具有较高立体位阻的烷基取代乙炔基溴底物同样能够良好地兼容 (3m–3n)。此外,作者发现,各类非活化的内烯 (3o–3w)同样能够有效地参与上述氢炔基化过程。接下来,作者进一步发现,苯乙烯底物同样能够顺利地完成相应的氢炔基化过程,并获得苄位炔基化产物 (3x–3k’)。同时,芳环中带有不同类型取代基的苯乙烯底物 (3z–3e’)以及杂环芳乙烯底物 (3f’,3g’)同样能够顺利地参与上述的氢炔基化反应过程。
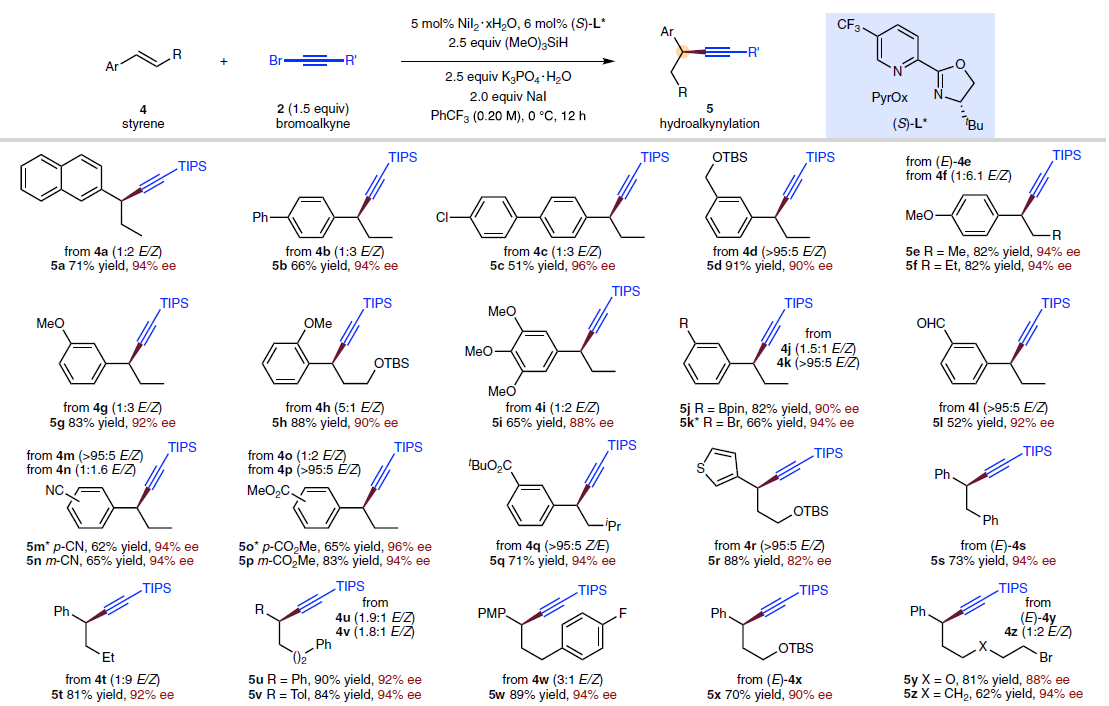
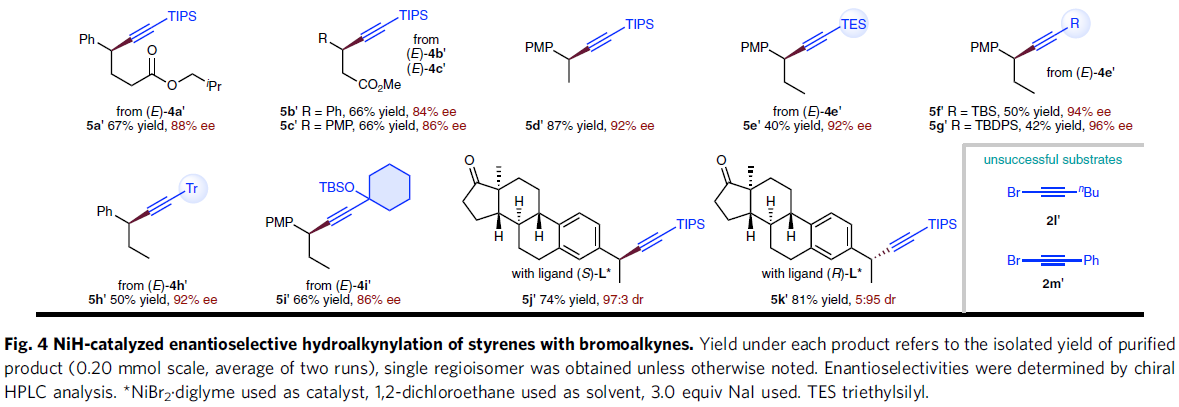
之后,通过对上述反应条件的进一步优化,该小组发现,在选择手性PyrOx配体(S)-L*时,能够有效地完成相应的对映选择性氢炔基化反应 (Fig. 4),并获得良好的产物收率与优良的对映选择性。同时,作者进一步观察到,芳环中具有不同基团取代的苯乙烯底物 (5a–5q),均能够顺利参与上述的不对称氢炔基化过程。值得注意的是,苯乙烯底物的芳环中存在能够进行进一步交叉偶联过程的取代基,例如氯 (5c)、溴 (5k)以及硼酸频哪醇酯基团 (5j)时,同样能够良好地兼容。同时,上述的标准反应条件对于β-位置具有不同类型取代基团的苯乙烯底物,均能够良好地进行兼容 (5r–5c’)。同时,研究表明,溴烷基取代基 (5y、5z)同样能够与上述反应体系有效地相容。并且,β-未取代的苯乙烯底物 (5d’、5j’、5k’)同样能够较好地采用上述的合成转化过程。接下来,作者对溴代炔的底物适用范围进行深入研究,实验表明,在β-位置中带有一系列不同立体位阻的取代基,例如,硅基以及烷基取代的乙炔基溴底物 (5e’–5i’),均能够良好地参与上述的氢炔基化过程。然而,立体位阻较小的烷基取代乙炔基溴底物 (2l’)与芳基取代的乙炔基溴底物 (2m’),在上述的标准反应条件下则易于分解,因而,无法有效地参与相应的氢炔基化反应过程。
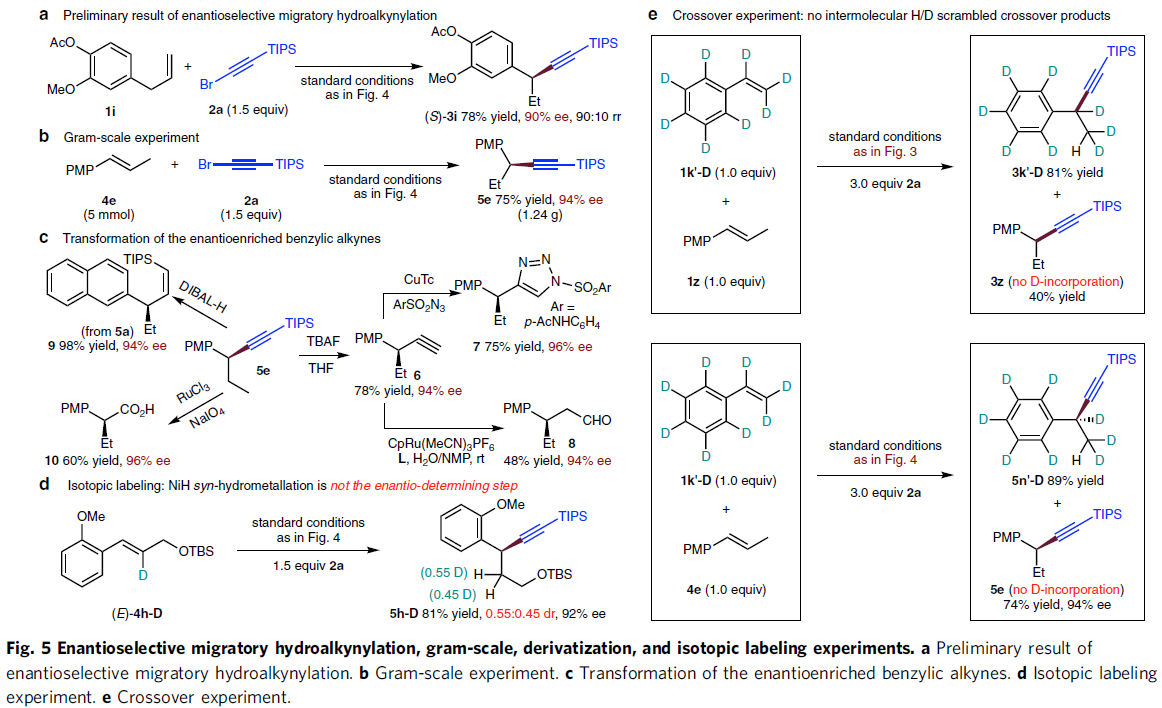
最后,作者对上述氢炔基化策略的实用性以及相关的反应机理进行研究 (Fig. 5)。首先,该小组发现,在上述标准条件下,采用3-芳基-1-丙烯(1i) 与2a底物,则能够进行相应的不对称迁移氢炔基化反应 (asymmetric migratory hydroalkynylation)过程,并获得产物(S)-3i,收率为78%,ee为90%,rr为90:10 (Fig. 5a)。并且,在4e与2a的克级实验中,同样能够获得具有较高反应收率与优良对映选择性的官能团化手性苄基炔产物 (5e, Fig. 5b)。同时,作者进一步发现,产物5e能够进行多种不同类型的衍生化反应 (Fig. 5c),例如,5e经历去甲硅烷基化过程,能够产生对映体富集的端炔 (6),继而通过后续的click环化与水合过程,分别获得产物7与8。同时,通过DIBAL-H对5e的类似产物5a进行的部分氢化(semi-hydrogenation) 反应,能够高度立体选择性地获得Z-烯基化合物 (9)。此外,通过5e中三键的氧化裂解过程,能够获得相应的手性羧酸10。
为初步研究相应金属氢化 (hydrometallation)过程的反应机理,作者进行相关的同位素标记实验 (isotope labeling experiment, Fig. 5d)研究。实验过程中,作者发现,在采用氘代的(E)-4h–D作为反应底物时,能够获得近似等量的两种非对映异构体 (0.55:0.45 dr),进而表明syn-金属氢化步骤并非相应的对映选择性决定步骤。并且,上述实验观察与作者最初提出的通过烷基镍 (III)中间体的快速均裂过程,最终形成苄基立体中心的机理一致。同时,通过后续的对映汇集过程(enantioconvergent process),镍 (II)中间体与苄基自由基结合,再度形成镍 (III)对映体 (Fig. 1c, ii)。 接下来,该小组进行相关的交叉实验研究。作者进一步研究表明,将1k´-D分别与1z与4e进行反应时,均未观察到分子间的H/D置乱交叉产物 (H/D scrambled crossover product)的形成。这一事实表明NiH/NiD配合物与苯乙烯之间的金属氢化步骤为不可逆过程 (Fig. 5e)。
总结
本文主要报道一种通过NiH催化剂促进的氢炔基化与不对称氢炔基化反应策略,进而成功完成一系列官能团化的苄位炔基化合物的构建。这一全新的氢炔基化策略,具有反应条件温和、底物应用范围广泛以及优良的官能团兼容性等优势。同时,该策略具有良好的合成应用价值。然而,对映选择性迁移氢炔基化过程的反应机理,则有待进一步研究。
















No comments yet.