
本文作者:杉杉
导读
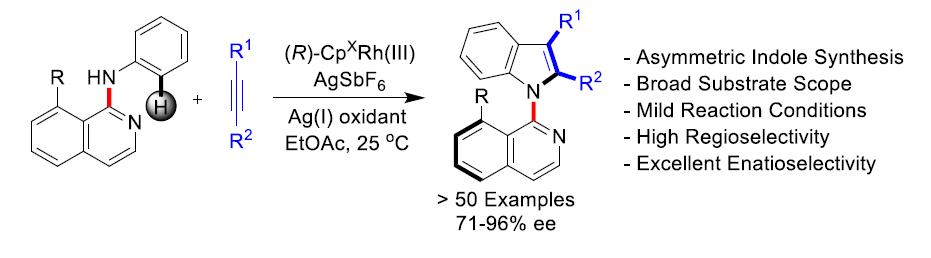
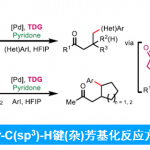
近日,陕西师范大学李兴伟课题组在Angew. Chem. Int. Ed.中发表论文,报道了通过铑(III)催化剂参与的苯胺(带有N-异喹啉基导向基团)C-H活化,并进一步与炔烃进行氧化[3+2]环合反应,从而完成一系列吲哚分子的阻转选择性(atroposelective)合成。该方法学具有温和的反应条件、高度的区域与对映选择性以及良好的官能团兼容性等优点。其中,陕西师范大学李兴伟教授、W. Fen博士以及重庆大学蓝宇教授(负责理论计算)为共同通讯作者。
Rhodium-Catalyzed Atroposelective Construction of Indoles via C-H Bond Activation
L. Sun, B. Liu, J. Chang, L. Kong, F. Wang, X. Li
Angew. Chem. Int. Ed. ASAP. DOI:10.1002/anie.202012932.
正文
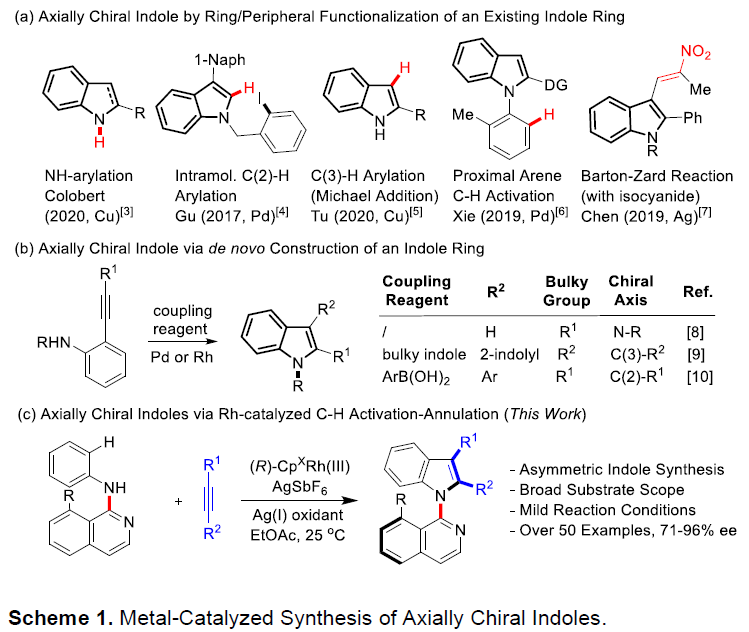
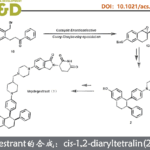
吲哚已经成为天然产物、药物分子与有机材料中广泛存在的结构单元。尤其是轴手性吲哚骨架作为普遍的手性砌块与配体,已经受到诸多课题组的关注。目前,关于轴手性吲哚的合成已有大量文献报道。例采用高价碘试剂、芳卤与醌以及金属催化剂,完成吲哚环的1、2与3-位的芳基化(Scheme 1a)。并且,与吲哚相连的相应芳环或烯基的官能团化,同样能够形成C-N或C-C手性轴(Scheme 1a)。此外,通过炔基取代苯胺的环化反应,同样能够以阻转选择性的方式实现吲哚骨架的从头构建(de novo construction)(Scheme 1b)。由此启发,Kitagawa等[1]报道了通过钯催化的炔烃环化反应,从而合成出一系列具有C-N轴手性的吲哚分子。同时,李兴伟课题组[2]将具有立体位阻的吲哚C-H活化与炔烃环化相结合,从而顺利完成2,3′-联吲哚的阻转选择性合成。Zhu等[3]通过选择芳基硼酸作为芳基化试剂,在Pd催化剂存在下,成功实现立体位阻炔烃的不对称Cacchi反应。然而,金属催化的手性吲哚的从头构建方法学仍然极少有文献报道。受到之前通过C-H活化策略构建联吲哚分子的启发,本课题组设想将这一策略应用于轴手性联芳分子的构建。进而设想采用如下两种反应设计的策略,即新的手性轴的从头构建以及基于分子中现有手性轴的动态动力学转化(dynamic kinetic transformation)。然而,多数动态动力学转化能够将邻位C-H键转化为不同的端基基团。Waldmann等[4]报道了采用手性JasCpRh(III)催化剂,进而完成带有侧链炔基的苯甲酰胺的分子内的氧化还原中性[4+2]环化反应。之后,本课题组[5]近期将Waldmann发展的这一成环方法学应用于分子间的反应过程。同时,Wang等[6]报道了通过Rh(III)催化,成功实现N-芳基吲哚满酮(N-arylindolinone)与炔烃之间的[2+ 2+2]碳环化反应([2+2+2] carboannulation)。然而,由于反应过程中采用较大立体位阻的导向基团或偶联试剂,因此,相关的文献报道极少,并且仅限于6,6-联芳化合物的合成。受到Fagnou等[7]开创的通过氧化偶联方法合成吲哚以及本课题组在2010年发展的氧化偶联合成外消旋吲哚分子[8]的启发,作者将苯胺的C-H活化方法学应用于N-异喹啉基吲哚啉(N-isoquinolylindoline)阻转异构体的合成(Scheme 1c)。尽管已有外消旋产物合成的相关报道,然而,对于其对映体选择性控制仍然具有挑战性,因为在C-N还原消除之前,需要对异喹啉导向基进行有效地构象控制。在此,陕西师范大学李兴伟课题组报道了铑(III)催化剂存在下进行的N-异喹啉基苯胺与炔烃之间的对映选择性[3+2]环合反应。
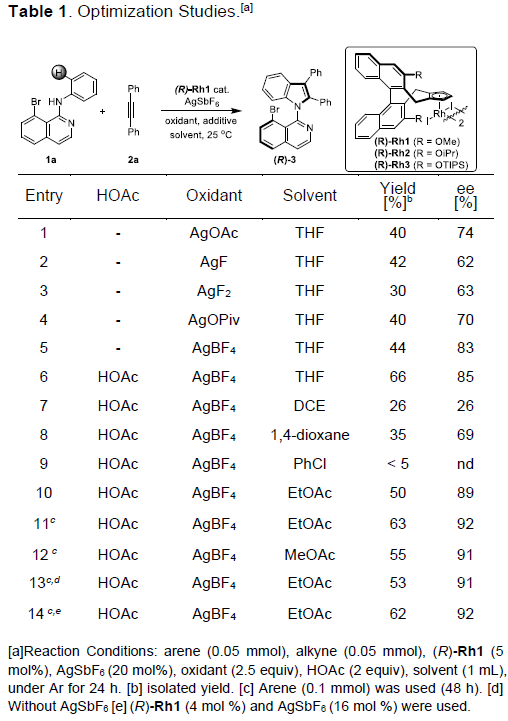
首先,作者采用苯胺1a与二苯乙炔2a作为模型底物,进行了相关反应条件的筛选(Table 1)。从而确定出反应的最佳条件为: (R)-Rh1作为催化剂, AgBF4作为氧化剂,乙酸乙酯作为反应溶剂,反应温度为25 oC,最终获得63%的反应收率与92%ee的目标产物(R)-3。
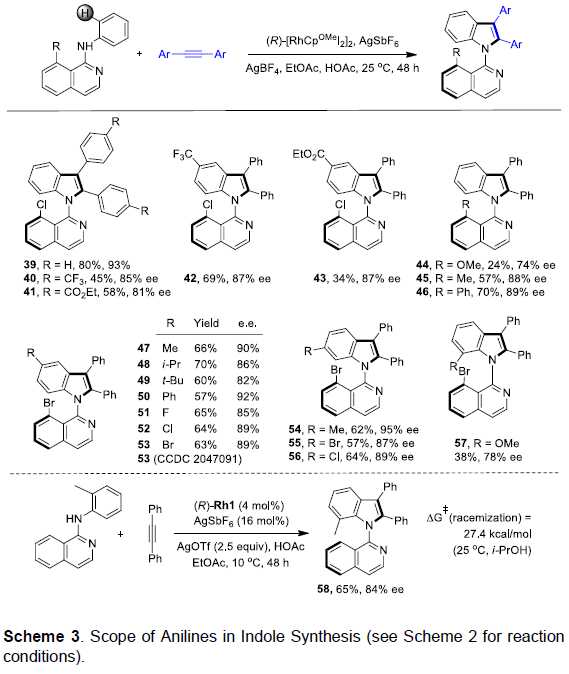
在获得上述最佳反应条件后,作者开始考察炔烃的底物适用范围(Scheme 2)。反应结果表明,对于芳环中不同位置存在各类吸电子基与供电子基的对称二芳基乙炔底物,均能够良好地兼容,并以较好的产率与优良的对映选择性获得相应的产物3–15(除苯环对位存在三氟甲基取代时,产物10的对映选择性略有下降外)。而且,在产物3(1 mmol)的放大反应实验中,作者发现其对映选择性与收率无明显下降。同时,实验发现,芳基二取代、杂芳基取代以及二烷基取代的炔烃底物,均能顺利进行反应,获得相应的产物16–20。然而,在采用烷基芳基炔底物时,实验发现,需要将氧化剂AgBF4替换为AgOPiv,才能够顺利完成相应转化,获得目标产物21–24。同样地,实验表明,选用醚-烷基与硅烷基醚-烷基底物时,同样可以获得产物25–30。然而,选择氯烷基取代的炔参与上述反应时,仍然可以获得高度的映选择性,而反应过程的区域选择性却较低(31)。此外,作者同样对1,3-烯炔的底物范围进行深入研究,然而,需要选用CH3SO3Ag作为氧化剂以及THF溶剂中进行反应,才能够获得具有优良对映选择性的环化产物32–38。

随后,作者对苯胺的底物适用范围进行了进一步扩展(Scheme 3)。研究表明,含有8-C-异喹啉取代基的苯胺底物,能够顺利与各类二芳基乙炔底物进行环化反应,获得相应的产物39–43。同样地,带有8-Me与8-Ph-异喹啉取代基的苯胺底物,能够获得高度对映选择性产物45与46,然而,带有8-OMe异喹啉取代基的苯胺底物(44),对映选择性与收率均较差。此外,作者进一步观察到,苯胺的对位与间位存在烷基、芳基以及卤素取代基时,均能够良好地与上述反应条件兼容,并获得相应的产物47–56。然而,对于邻甲氧基苯胺底物(57),则观察到对映选择性与收率显著下降。值得注意的是,采用AgOTf作为氧化剂,能够在温和的条件下使异喹啉基邻甲基苯胺底物与二苯基乙炔发生上述环化反应,获得较高收率与对映选择性的产物58,尽管外消旋化的能垒较低。
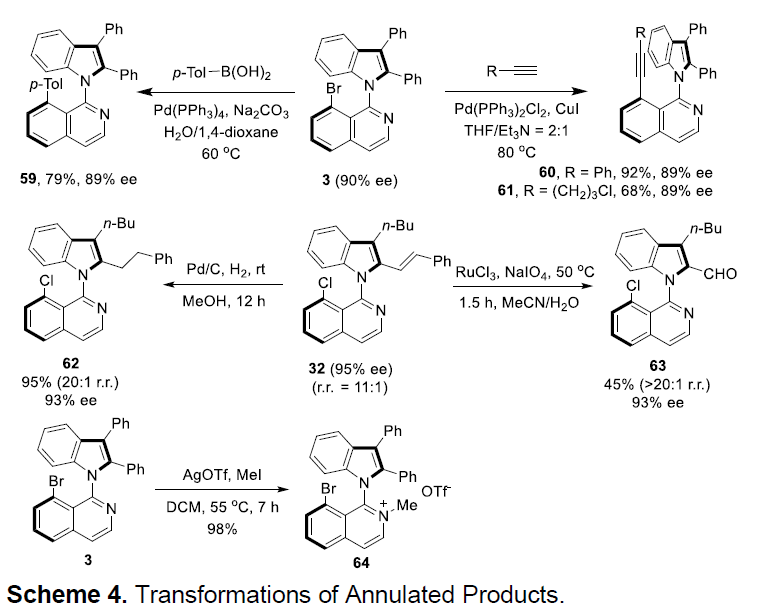
接下来,为了证明该反应的实用性,作者对相应目标产物进行一系列衍生化反应(Scheme 4)。首先,将产物3与p-TolB(OH)2进行Suzuki偶联,获得产物59。之后,发现3同样能够与不同炔烃进行Sonogashira偶联反应,获得产物60与61。同时,作者发现32经过氢化,能够获得产物62,而经过C=C氧化断裂,能够获得产物63。最后,发现将3进行甲基化反应,能够获得较高收率的异喹啉鎓盐64。
最后,作者对反应的机理进行了深入研究(Scheme 5)。首先,在H/D交换实验中,作者观察到1a与CD3COOD之间在有2a或无2a存在的条件下进行的H/D交换均能够使苯胺的邻位出现明显的氘代。这表明C-H活化为可逆过程(Scheme 5a)。同时,在平行的KIE实验中,观察到较大的kH/kD数值,kH/kD= 4.9。表明反应决速步骤中存在C-H的断裂(Scheme 5b)。接下来,作者通过DFT计算对反应过程中的不对称诱导方式进行研究。结果表明,反应可能涉及四种可能的过渡态(TS-R-1,TS-R-2,TS-S-1和TS-S-2)(Scheme 5c)。形成R型与S型产物的能量最低的还原消除机理路径分别由过渡态TS-R-1与TS-S-2决定。同时,过渡态TS-R-1比TS-S-2的活化自由能要低5.8 kcal/mol,这与观察到的R型选择性相一致。此外,在过渡态TS-S-2中,由于手性配体中的甲氧基与苯乙烯骨架的苯基之间存在一定的立体排斥,因而使苯乙烯骨架中苯基的扭转二面角大于TS-R-1(分别为147.4o与115.3o)。

![]()
总结
本文主要介绍了通过铑(III)催化剂参与的苯胺(带有N-异喹啉基导向基团)C-H活化,并进一步与炔烃进行具有高区域与对映选择性的氧化[3+2]环合反应,从而顺利完成一系列N-异喹啉基吲哚衍生物的阻转选择性合成。与之前报道的动态动力学转化相反,上述[3+2]环加成策略通过轴手性氮原子作为反应位点。此外, DFT研究表明,反应中的不对称诱导过程可能涉及两种非对映异构烯基Rh(III)物种的还原消除。













No comments yet.