
本文作者:石油醚
导读
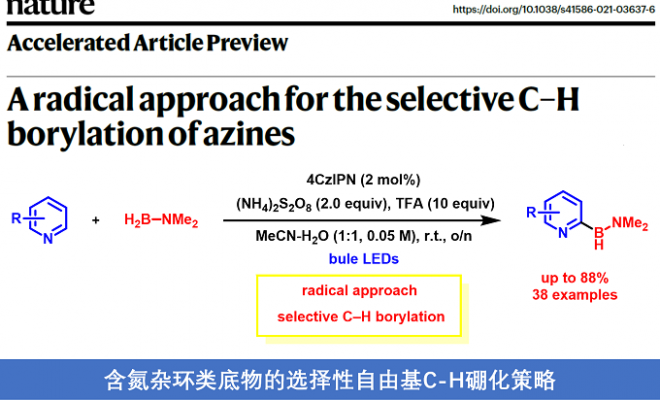
自上世纪70年代Suzuki偶联反应的研究被报道以来,针对芳香类有机硼试剂的研究越来越深入,目前已经成为有机合成当中十分普遍的一种合成砌块。硼类试剂尤其是芳香类硼试剂在有机合成、药物化学、功能材料等领域的应用已极其广泛,而且随着时间的推移还会更加重要。目前,过渡金属催化芳香族与杂芳香族衍生物C-H活化构建C-B键是十分有效,但是对于含氮芳杂环化合物(吡啶、嘧啶和喹啉等)以及许多医药和农药产品的关键成分的适用性很小。近日,曼彻斯特大学Daniele Leonori教授课题组在Nature发表了题为“A radical approach for the selective C–H borylation of azines”的论文,该课题组在蓝光条件下,利用光催化剂4CzIPN与商业可得胺-硼烷制备出稳定且高反应性的胺-硼自由基,进而有效实现了含氮杂环类底物的选择性C-H硼化反应。该策略制备了稳定且高反应活性的胺-硼自由基,并为构建含氮杂环化合物C-B键提供了新的策略。
Kim, J. H.; Constantin, T.; Simonetti, M.; Llaveria, J.; Sheikh, N. S.; Leonori, D*.
Nature 2021(Just accepted), doi:10.1038/s41586-021-03637-6.
正文
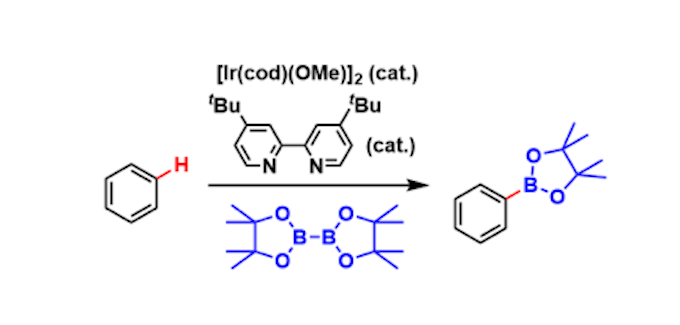
芳香类有机硼试剂是合成化学最基础的组成部分之一,Suzuki以芳基硼试剂作为偶联底物获得了诺贝尔化学奖。除此之外,芳香类有机硼试剂还在氧化,Chan-Lam交叉偶联,氟化和同系化等过程中充当底物,用以构建新的化合物。近几十年,芳香类有机硼试剂C–H活化直接芳烃硼化的方法是先制备芳基卤化物,而后通过格式试剂或者Pd催化实现Miyaura 硼化(图1)。该领域最大的突破为开发了金属Ir / Rh催化(B2(pin)2/HB(pin)实现 C–H活化直接芳烃硼化(图1)。以上方法因其消除芳香族化合物的预官能化,其在学术界和工业界被广泛使用。尽管芳族衍生物的C–H活化直接芳烃硼化是一种普遍的策略,但其选择性不好,含氮底物的适应性不好、C2位取代氮杂环产物不稳定、反应位点单一等缺点,严重阻碍了其在N–杂芳族化合物药物和农用化学品的构建方面的应用。Daniele Leonori教授在研究开发一种全新的含氮杂环化合物硼化反应策略时,希望通过Minisci反应途径将B-官能团化引入烷基。目前,构建C(Sp2)-B键策略常常是使用硼试剂作为亲电子试剂(格氏试剂和C-H硼化)或亲核试剂(Miyaura硼化),而“ Minisci式”方法将需要以硼为中心的自由基作为中间体。这些反应性中间体在合成化学中的代表性不高,但它们有可能通过选择性C-H硼化反应,实现含氮杂环类底物有机硼化合物的制备。Daniele Leonori教授认为,硼基自由基前体的发现对于成功实现含氮杂环选择性自由基C-H硼化策略至关重要,该硼基自由基前体需要同时解决三个主要因素。如(i)温和条件下使用廉价易得的硼化试剂,制备应为稳定和高反应活性的硼基自由基前体;(ii)由于Minisci反应得益于亲核烷基自由基的使用,B取代模式对于产生具有相关亲核性的中间物种很重要;(iii)最后,硼化的含氮杂环产物是稳定的,可以进一步官能团化的功能。
图 1 传统的选择性C-H硼化反应
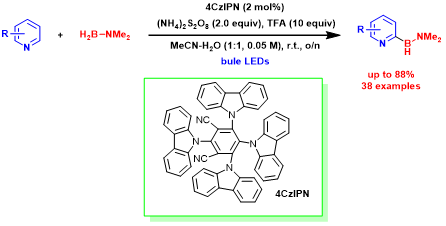
Daniele Leonori教授考虑到上述的方面后,使用聚合物前体和储氢材料廉价易得的叔胺与BH3形成的路易斯碱加和物(胺-硼烷)代替通用的NHC-硼烷,这主要是因为自由基离域到NHC环中会降低亲核特性,这可能会妨碍Minisci型化学的应用。尽管胺-硼烷在有机合成中的应用受到限制,但Roberts的开创性研究吸引Daniele Leonori了的注意力,该研究表明胺配位硼烷发生HAT过程生成稳定和高活性的硼自由基,来促使相应的物种具有好的亲核性(图 2)。
基于以上,Daniele Leonori教授课题组在蓝光条件下,利用光催化剂4CzIPN与商业可得胺-硼烷制备出稳定且高反应性的胺-硼自由基,高进而有效实现了含氮杂环类底物的选择性C-H硼化反应。Daniele Leonori教授小组对选择性C-H硼化反应做了详细机理描述。该过程是:蓝光条件下,光催化剂(PC)与过硫酸盐A发生单电子转移(SET)并生成相应的硫酸根自由基阴离子B。随后,胺-硼烷会受到O-自由基B的影响,发生HAT过程制备出稳定并且高反应活性的胺-硼自由基C,胺-硼自由基C与质子化的3-H+的C2位发生Minisci加成反应获得了自由基中间体E,这样,我们就通过简单的自由基加成反应(作为C-B键的构建过程)代替了从含氮杂环-有机金属中间体的还原消除过程。最后,经过光催化剂(PC•+)的SET过程,或者通过过硫酸盐A的直接SET(自由基链式反应)过程促使自由基中间体 E去质子化形成3-H+,完成催化循环(图 2)。该过程需要化学计量的Brønsted酸协同催化反应:(i)促进SET过程的发生降低A含量;(ii)增加B的亲电性,促进HAT过程的发生;(iii)通过C提高含氮杂环对Minisci硼试剂的亲电性;(iv)将所得的胺-硼烷产物3 不在发生B中HAT过程。实际上实际上,实际上,根据计算,3中的B-H键要弱于2中的B-H键,但N-质子为产物提供了动力学稳定性,防止3进一步的HAT过程,从而导致产物分解。关于关键的C-B形成步骤,将等电子的与新戊基F添加到1-H +中, C在热力学和动力学上都更加容易发生加成反应。最后,Daniele Leonori教授发现,与F相比胺-硼自由基C拥有的更高的亲核性,在硼化的过渡态中会产生强大且稳定的极性效应,并且硼基自由基的空间位阻会进一步促进这种效应。
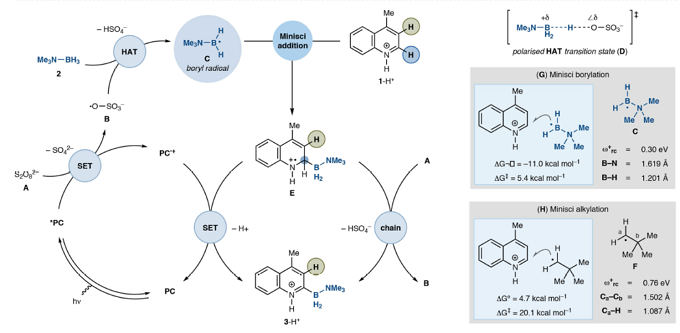
图 2含氮杂环化合物的选择性C-H硼化反应机理
令作者欣喜的是,室温条件下,在蓝光照射下,使用有机染料4CzIPN作为光催化剂,TFA作为布朗斯特酸和(NH4)2S2O8作为氧化剂,以CH3CN-H2O溶剂就可以实现含氮杂环喹啉1与胺-硼烷2的选择性自由基C-H硼化反应(图3)(反应优化和对照实验见SI)。在此条件下,1和2在几克规模上高产率获得3。 二在不存在4CzIPN的情况下,也可以获得3,但是这个条件不能转化为下面的的底物范围。3产物可以通过重结晶获得,室温条件下稳定6个月。
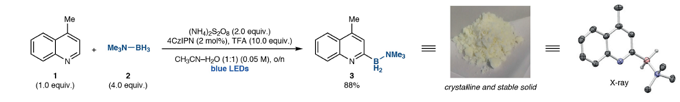
图 3含氮杂环化合物的选择性C-H硼化反应
在获得上述最佳反应条件后,作者开始对不同结构的含氮杂环底物(图 4),如C4取代的喹啉、C2位未被取代的吡啶(15-17)、取代的哒嗪(20-27)、嘧啶类(28-30)、azaindazole(31-32)、咪唑并[1,2-b]哒嗪(33)、噻吩并[3,2-d]嘧啶(34)和苯并噻吩(35)等众多含氮杂环化合物都可以的发生选择性C-H硼化反应,并对羟基、氨基、卤素等官能团就有额外的耐受性。选择性C-H硼化反应的策略,也可以很好地应用到药物分子的后期修饰过程中(36-41),令人高兴的是,基于(异)喹啉的Agrochemical的硼化,辛可尼定(克级规模)和法舒地尔(血管扩张剂)也可以很好地发生反应,证明该反应与苄醇,叔胺,烯烃和磺酰胺官能团具有额外的耐受性。结构复杂的抗真菌药伏立康唑在C6(40,克级规模),对其5-氟嘧啶核也可以发生选择性硼化。最后,Daniele Leonori教授小组能够使抗病毒药物泛昔洛韦存在不稳定的O-醋酸酯基团的情况下使嘌呤核心骨架的C6位发生硼化反应。(41)。
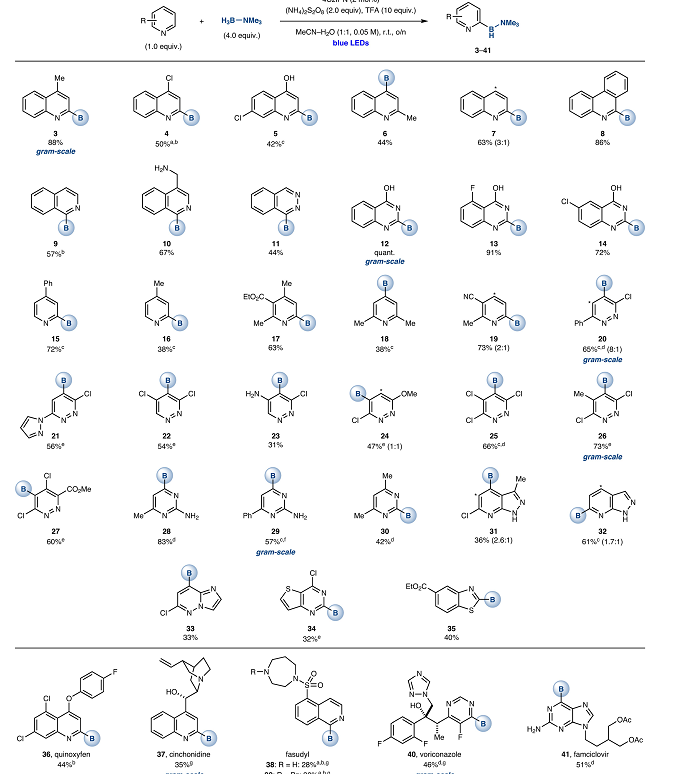
图 4 底物拓展
最后,为了体现胺-硼自由基选择性C-H硼化策略的潜力,含氮杂环有机硼化合物进一步官能团化是将其纳入有机硼化合物领域至关重要的一步。作者对胺-硼自由基选择性C-H硼化获得产物6为模板底物,成功实现了含氮杂环有机硼化合物后期的官能团化,如氧化、Suzuki-Miyaura交叉偶联、Chan-Lam胺化以及Chan-Lam醚化反应(图5),后期对不同含氮杂环的有机硼化合物具有很好地适用性,体现了胺-硼自由基选择性C-H硼化策略的潜力。
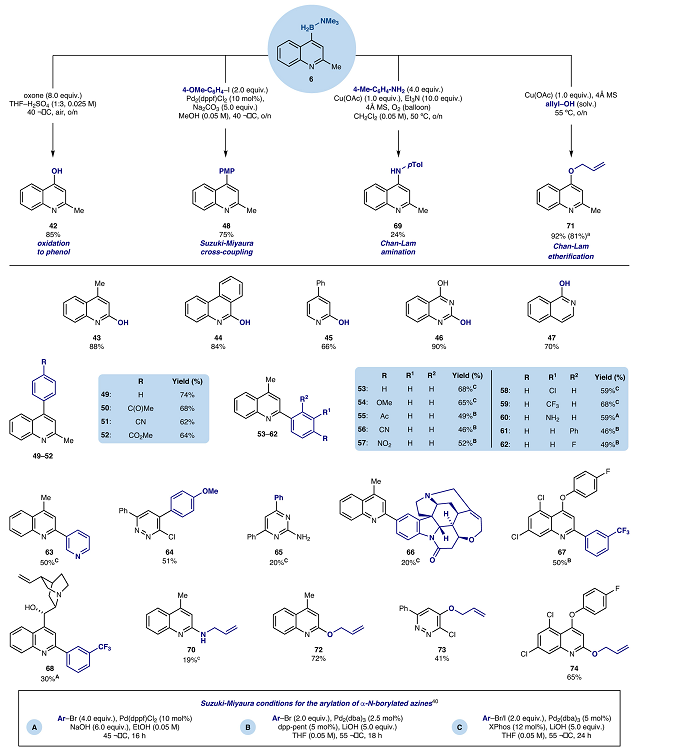
图 5含氮杂环有机硼化合物后期修饰
Daniele Leonori教授课题组在蓝光条件下,利用光催化剂4CzIPN与商业可得胺-硼烷制备出稳定且高反应性的胺-硼自由基,进而有效实现了含氮杂环类底物的选择性C-H硼化反应。该策略制备了稳定且高反应活性的胺-硼自由基,并为构建含氮杂环化合物C-B键提供了新的策略,获得产物稳定,对其可以使用氧化、Suzuki-Miyaura交叉偶联、Chan-Lam胺化以及Chan-Lam醚化后期官能团修饰。该策略的成功实现,在药物化学和硼酸化学领域有更加广泛的意义。


















No comments yet.