
本文作者:杉杉
导读
近日,Vienna大学N. Maulide课题组在J. Am. Chem. Soc.中发表论文,报道一种通过酰胺化合物的N-脱氢 (N-dehydrogenation)反应策略,进而顺利实现一系列酰基烯胺 (enamide)分子的构建。在该策略的设计中,选择LiHMDS与Tf2O结合,并将其同时作为亲电活化剂 (electrophilic activator)与氧化剂。同时,该方法学具有实验操作简便、底物应用范围广泛等优势。并且,作者通过对相应酰基烯胺产物的后期衍生化实验,进一步阐明上述N-脱氢策略的合成应用价值。
Direct Synthesis of Enamides via Electrophilic Activation of Amides
P.Spieß, M.Berger, D. Kaiser, N. Maulide J.Am. Chem. Soc.ASAP. doi: 10.1021/jacs.1c04363.
正文
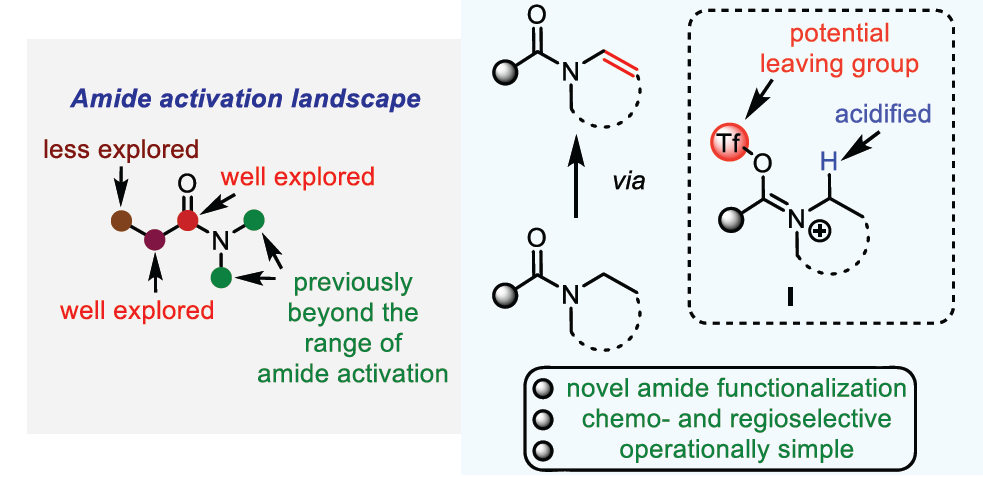
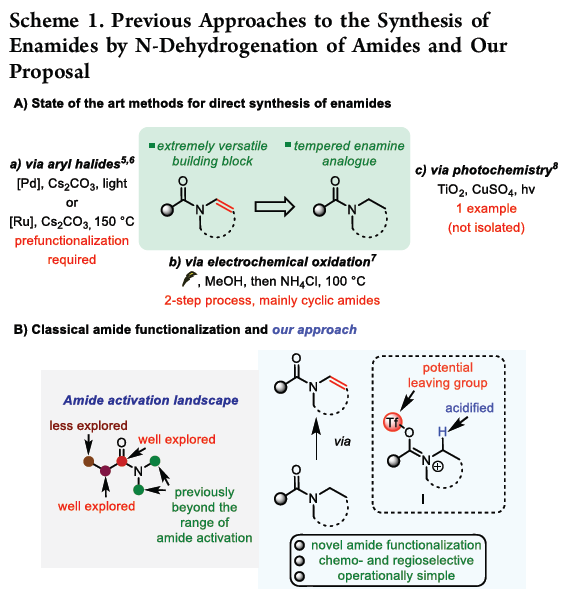
烯胺为有机合成工具箱 (organic synthetic toolbox)中基本的合成砌块,通常表现出优良的亲核反应活性。同时,由于烯胺化合物较为独特的反应活性,并且,极易水解,因而实验操作较为繁琐。然而,与烯胺类化合物相比,酰基烯胺 (enamide)化合物则具有良好的湿气稳定性与适宜的反应活性,因而,能够有效地替代烯胺分子,并应用于一系列合成转化策略的研究。而且,近年来,酰基烯胺化合物已经广泛用于过渡金属催化、光化学以及不对称催化反应的相关研究[1]。在酰基烯胺化合物的合成中,通常需要采用预官能团化 (prefunctionalization)的反应底物[2]。而相应酰胺底物的N-脱氢反应 (N-dehydrogenation)方法学,则成为酰基烯胺化合物构建中,最为直接的反应策略 (Scheme 1A)。例如,Gevorgyan课题组[3]报道采用光诱导条件下,通过钯催化的预官能团化的2-碘苯甲酰胺 (prefunctionalized 2-iodobenzamides)底物的脱氢策略,完成一系列酰基烯胺分子的构建。同时,Morandi课题组[4]发现,在钌催化条件下,同样能够实现预官能团化2-碘苯甲酰胺的脱氢过程。并且,同样有文献[5]报道通过电化学氧化策略进行的N-脱氢反应。其中,酰胺底物大多为环酰胺,在反应过程中转化为半缩醛胺甲醚 (hemiaminal methyl ether),并通过酸性条件下的进一步水解,获得相应的酰基烯胺产物[6]。此外,通过光媒介以及二氧化钛与铜(II)盐存在的条件下,N-乙酰基吡咯烷的一步氧化策略更加具有吸引力[7]。然而,产物仅能够通过波谱分析检测出,并未进行分离 (Scheme 1A)。此外,设计一种无需将底物进行预官能团化处理,并能够有效兼容N-环状以及非环酰胺底物的反应策略,仍然具有较大的挑战。近年来,亲电酰胺活化 (electrophilic amide activation)策略[8]由于能够有效地解决酰胺底物亲电性较差的问题而备受关注。尤其采用三氟甲磺酸酐与相应吡啶碱的结合,能够有效地实现羰基位置以及羰基的α-官能团化 (Scheme 1B) [9]。然而,对于酰胺底物的N-官能化,目前尚未有相关的文献报道。基于上述研究背景,作者设想,能否通过亚胺三氟甲磺酸盐 (iminium triflate)中间体 I的形成,顺利实现酰胺类化合物N-脱氢反应过程,进而完成酰基烯胺分子的构建。同时,作者进一步设想通过亚胺三氟甲磺酸盐中间体 I的形成,能够使氮原子的α-H酸性增强,进而促进后续去质子化过程 (采用较强的非亲核碱 (nonnucleophilic base))的有效进行。基于上述设想,作者成功设计出一种通过亲电酰胺活化的方式,将各类酰胺化合物转化为酰基烯胺分子的方法学策略。
首先,作者采用1a作为模型底物,进行了相关脱氢反应条件的优化筛选 (Table 1)。最终确定最佳的反应条件为:采用4.8 eq. LiHMDS与2.4eq. 三氟甲磺酸酐 (Tf2O)结合,乙醚作为反应溶剂,反应温度为-94 oC,进而获得89%收率的酰基烯胺产物2a。

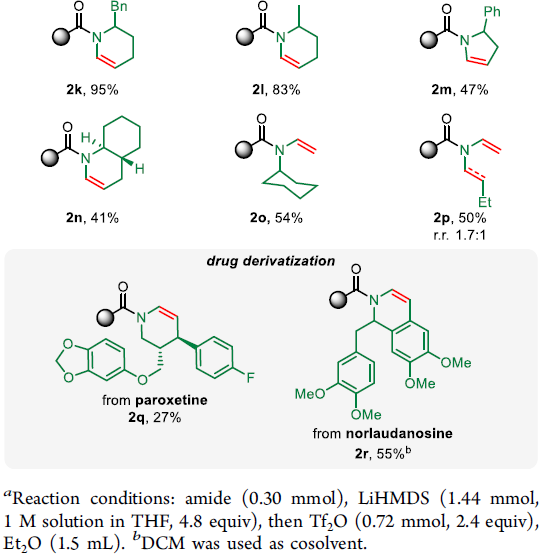
在上述最佳反应条件下,作者首先对相应N-烷基酰胺底物的应用范围进行考察 (Scheme 2)。研究发现,具有不同环尺寸的酰胺 (2a–2c)、杂原子取代的酰胺 (2d)、双环酰胺 (2e)以及吗啉与哌嗪衍生的酰胺 (2f与2g),均能够顺利地参与上述反应。同时,实验过程中作者发现,非环酰胺同样能够与上述反应体系良好地兼容,并获得E-酰基烯胺2h–2j。此外,作者发现,对于非对称酰胺底物,反应优先选择N-取代基中立体位阻较小的位置进行相应的N-脱氢反应,并获得产物2k–2o。对于同时带有乙基与丁基取代的酰胺底物,采用上述反应条件,同样能够获得中等程度的区域选择性 (2p)。值得注意的是,这一策略同样能够成功应用于药物相关分子的后期修饰,例如2q与2r。

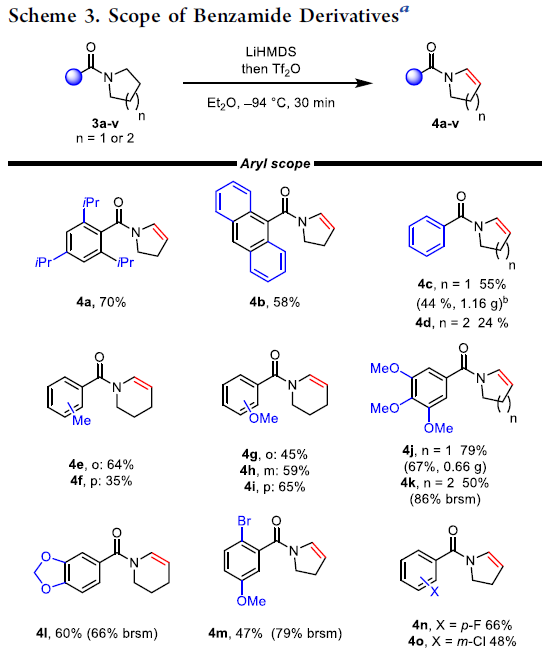
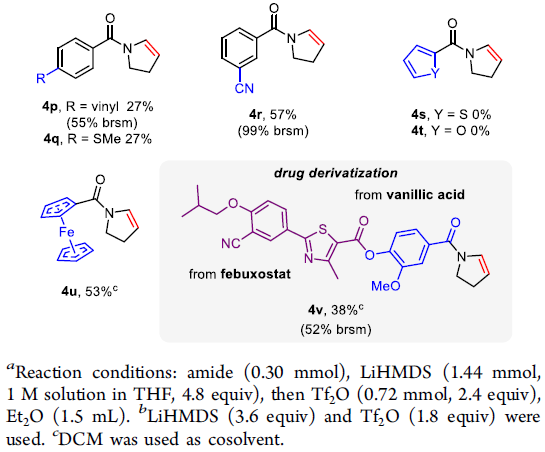
之后,作者对相应苯甲酰胺衍生物的底物适用范围进行考察 (Scheme 3)。研究发现,芳环中具有较大立体位阻基团取代的苯甲酰胺底物,均能够顺利参与上述反应,并获得相应目标产物4a与4b。对于芳环中无取代基存在的苯甲酰胺底物,则获得较低收率的产物4c与4d。同时,研究发现,对于芳环中不同位置带有甲基 (4e,4f)取代的苯甲酰胺底物,在邻位取代时,反应收率最高。而芳环中不同位置带有甲氧基 (4g–4i)取代的苯甲酰胺底物,则在对位取代时,反应收率最高。其次,芳环中带有其它富电子基团 (4j–4l)以及卤素基团 (4m–4o)取代的芳甲酰胺底物,同样能够有效地完成相应的N-脱氢反应。同时,上述的N-脱氢反应,具有良好的官能团兼容性。然而,噻吩甲酰胺与呋喃甲酰胺 (3s与3t)却无法与上述反应条件良好地兼容。值得注意的是,上述脱氢策略能够成功应用于二茂铁衍生的酰基烯胺 (4u)以及部分药物分子 (4v)的合成。
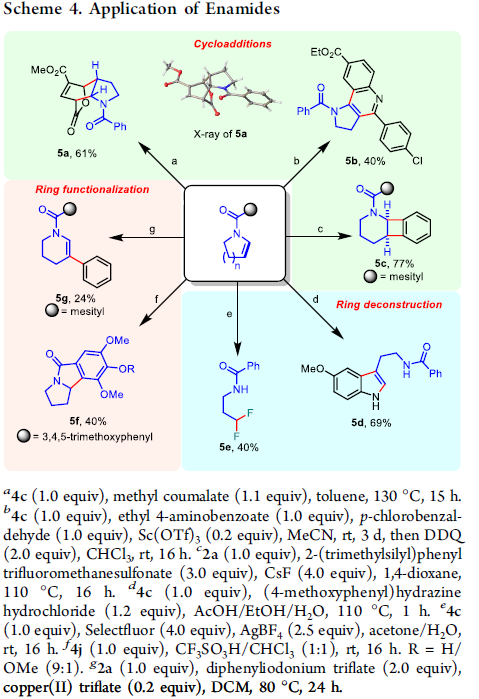
为了进一步阐明上述反应的合成应用价值,接下来,作者进行一系列相关的目标产物官能团化实验 (Scheme 4)。首先,作者发现,酰基烯胺产物能够较为容易地参与IEDA (inverse electron-demand Diels-Alder reaction) 与 [2+2] 环加成反应过程,并分别获得相应产物5a–5b以及5c。并且,酰基烯胺产物同样能够有效地进行相关的开环过程,并获得相应开环产物 5d与5e。此外,研究发现,在酸性条件下,酰基烯胺产物能够较好地完成Nazarov环化过程,并获得良好收率的三环内酰胺分子 5f。最后,在铜催化条件下,能够较为容易地完成酰基烯胺产物的β-芳基化反应,并获得中等收率的芳基化产物 5g。
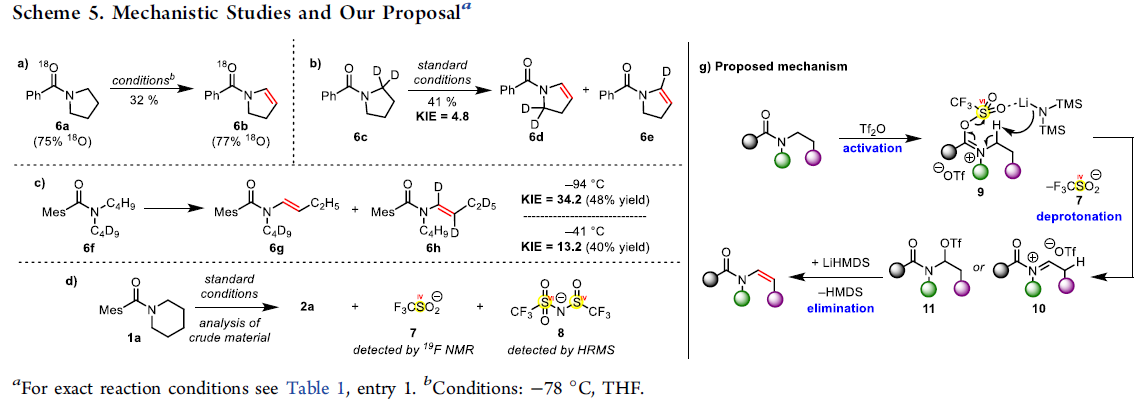
最后,作者对反应机理进行深入研究 (Scheme 5)。首先,作者发现,采用富含18O的酰胺 (18O-enriched amide)底物 6a,在THF溶剂中,-78 ℃下进行反应,能够获得酰基烯胺化合物 6b,并且,18O标记未发生改变。这一结果表明反应机理中涉及亲电酰胺活化过程。同时,作者进一步发现,采用氘代底物 6c与6f在标准条件下进行反应时,环状底物 6c的KIE值 (6d:6e)为4.8,非环状底物6f的KIE值 (6g:6h)为34.2。上述事实表明,决速率步骤中涉及N-α–H的攫取。接下来,作者观察到,在提升反应温度 (-41℃)时,6f的KIE值降为13.2。此外,通过1a反应时,相关反应混合物的19F-NMR分析,能够检测出三氟甲磺酸盐 (7)的存在。并且,通过HRMS,能够进一步检测出化合物 8 (参阅SI) 的存在。更为重要的是,通过氘代实验 (deuteration experiment)表明,通过碱并无法直接攫取相应的的N-α–氢,由此能够排除去质子化-三氟甲磺酰化机理 (deprotonation/triflation mechanism, 参阅SI)。基于上述的实验观察,作者提出一种可能的反应机理。首先,酰胺底物通过Tf2O的活化,形成亚胺三氟甲磺酸盐,之后,通过LiHMDS对亚胺三氟甲磺酸盐中α-氢的去质子化/消除过程,移除三氟甲磺酸阴离子7,形成中间体10或11。最后,10或11经历进一步的消除过程,最终获得酰基烯胺产物。
总结
Vienna大学N. Maulide课题组报道一种采用LiHMDS与Tf2O体系,成功实现一系列酰胺化合物N-脱氢反应,进而完成各类酰基烯胺分子的构建。值得注意的是,这一策略为构建N-环状与非环酰基烯胺分子的首例较为通用的一步合成路线。同时,反应过程中,无需进行底物的预官能化步骤。此外,通过该方法学在药物相关的分子后期修饰、酰基烯胺衍生物的环加成以及开环反应等方面的应用研究,进一步阐明该策略具有良好的合成应用价值。
参考文献
[1] (a) F. Beltran, L. Miesch, Synthesis 2020, 52, 2497. doi: 10.1055/s-0040-1707403.(b) M. Wang, Chem. Commun. 2015, 51, 6039. doi: 10.1039/C4CC10327K.
(c) T. Courant, G. Dagousset, G. Masson, Synthesis 2015, 47, 1799. doi: 10.1055/s-0034-1378706.
(d) K. Gopalaiah, H. B.Kagan, Chem. Rev. 2011, 111, 4599. doi: 10.1021/cr100031f.
(e) R. Matsubara, S. Kobayashi, Acc. Chem. Res. 2008, 41, 292. doi: 10.1021/ar700098d.
(f) D. R. Carbery, Org. Biomol. Chem. 2008, 6, 3455. doi: 10.1039/B809319A.
[2] (a) B. M. Trost, J. J. Cregg, N. Quach, J. Am. Chem. Soc. 2017, 139, 5133. doi: 10.1021/jacs.7b00564.(b) L. Wang, C. Liu, R. Bai, Y. Pan, A. Lei, Chem. Commun. 2013, 49, 7923. doi: 10.1039/C3CC43875A.
(c) Y. Bolshan, R. A. Batey, Angew. Chem. Int. Ed. 2008, 47, 2109. doi: 10.1002/anie.200704711.
[3] P. Chuentragool, M. Parasram, Y. Shi, V. Gevorgyan, J. Am.Chem. Soc. 2018, 140, 2465. doi: 10.1021/jacs.8b00488. [4] L. Huang, A. Bismuto, S. A. Rath, N. Trapp, B. Morandi, Angew. Chem. Int. Ed. 2021, 60, 2. doi: 10.1002/anie.202015837. [5] (a) T. Shono, Y. Matsumura, K. Tsubata, Y. Sugihara, S. Yamane, T. Kanazawa, T. Aoki, J. Am. Chem. Soc. 1982, 104, 6697. doi: 10.1021/ja00388a037.(b) S. S. Libendi, Y. Demizu, Y. Matsumura, O. Onomura, Tetrahedron 2008, 64, 3935. doi: 10.1016/j.tet.2008.02.060.
[6] T. Golub, J. Y. Becker, Electrochim. Acta. 2016, 205, 207. doi: 10.1016/j.electacta.2016.04.074. [7] J. W. Pavlik, S. Tantayanon, J. Am. Chem. Soc. 1981, 103, 6755. doi: 10.1021/ja00412a043. [8] (a) N. A. Weires, E. L. Baker, N. K. Garg, Nat. Chem. 2016, 8, 75. doi: 10.1038/nchem.2388.(b) N. F. Hie, T. K. F. Nathel, E. L. Shah, E. L. Baker, X. Hong, Y. Yang, P. Liu, K. N. Houk, N. K. Garg, Nature 2015, 524, 79. doi: 10.1038/nature14615.
(c) A. Wang, C. Yu, T. Chen, Y. Liu, P. Huang, Org. Lett. 2018, 20, 999. doi: 10.1021/acs.orglett.7b03943.
(d) D. Kaiser, A. Bauer, M. Lemmerer, N. Maulide, Chem. Soc. Rev. 2018, 47, 7899. doi: 10.1039/C8CS00335A.
(e) D. Kaiser, N. Maulide, J. Org. Chem. 2016, 81, 4421. doi: 10.1021/acs.joc.6b00675.
[9] (a) M. Movassaghi, M. D. Hill, O. K. Ahmad, J. Am. Chem. Soc. 2007, 129, 10096. doi: 10.1021/ja073912a.(b) M. Movassaghi, M. D. Hill, Org. Lett. 2008, 10, 3485. doi: 10.1021/ol801264u.
(c) P. Adler, C. J. Teskey, D. Kaiser, M. Holy, H. H. Sitte, N. Maulide, Nat. Chem. 2019, 11, 329. doi: 10.1038/s41557-019-0215-z.
(d) M. Leypold, K. A. D´Angelo, M. Movassaghi, Org. Lett. 2020, 22, 8802. doi: 10.1021/acs.orglett.0c03160.
(e) C. R. Gonçalves, M. Lemmerer, C. J. Teskey, P. Adler, D. Kaiser, B. Maryasin, L. González, N. Maulide, J. Am. Chem. Soc. 2019, 141, 18437. doi: 10.1021/jacs.9b06956.
(f) K. Xiao, A. Wang, P. Huang, Angew. Chem. Int. Ed. 2012, 51, 8314. doi: 10.1002/anie.201204098.
(g) I. L. Baraznerok, V. G. Nenajdenko, E. S. Balenkova, Tetrahedron 2000, 56, 3077. doi: 10.1016/S0040-4020(00)00093-4.














No comments yet.