
本文作者:杉杉
导读
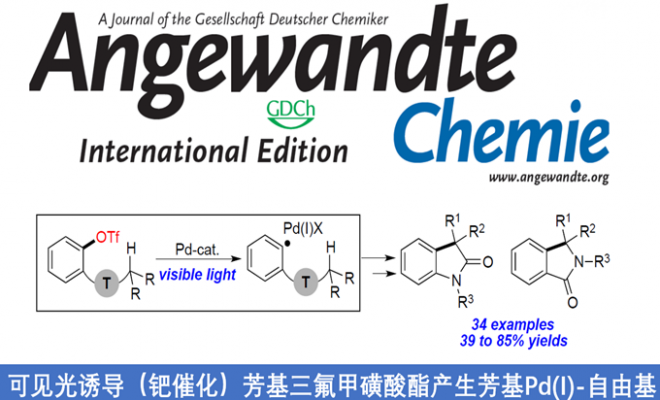
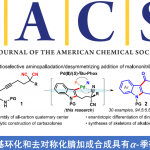
近日,伊利诺伊大学(University of Illinois)的Vladimir Gevorgyan教授团队在德国应化杂志发表论文,提出了可在温和的可见光诱导条件下,通过Pd催化实现酰胺分子内C-H芳基化反应。该方法通过C(sp2)-O键的断裂产生杂芳基Pd(I)-自由基中间体,涉及1,5-HAT/分子内环化/芳香化等过程,并获得多种具有价值的羟吲哚和异吲哚啉-1-酮的衍生物。此外,传统Pd催化条件下对碱敏感的官能团(如酮和酯)也可通过此方法来制备。
Visible Light-Induced Palladium-Catalyzed Generation of Aryl Radicals from Aryl Triflates
Maxim Ratushnyy, Nikita Kvasovs, Sumon Sarkar, Vladimir Gevorgyan
Angew. Chem. Int. Ed. ASAP DOI: 10.1002/anie.201915962
正文
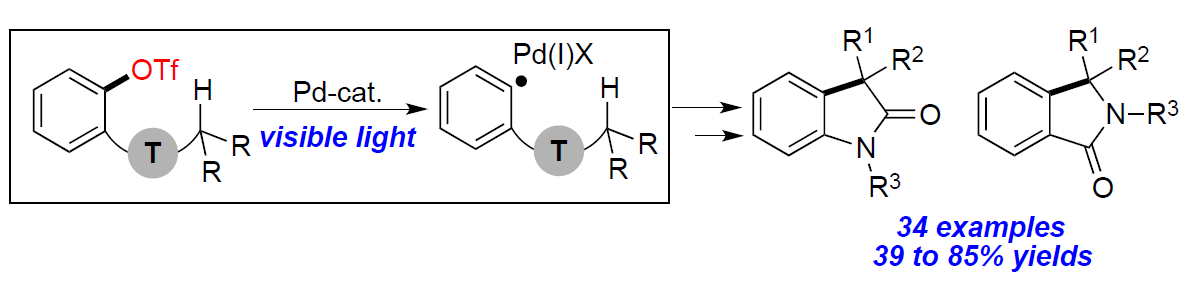
芳基卤化物作为各种Pd催化反应中重要的底物,这些转化常常通过双电子氧化加成形成Pd(II)中间体。最近,一种涉及通过单电子转移(SET)的新方法,可从光激发的Pd(0)络合物与有机卤化物生成Pd(I)中间体,作为Pd催化领域而迅速发展。最近,本课题组报道了可见光诱导的钯催化芳基碘化物C(sp2)-I键断裂,形成芳基杂化的Pd(I)-自由基中间体A(Scheme 1a),后者经历了选择性的1,5-氢原子移位步骤(1,5-HAT)获得中间体B,随后进行了β-H消除,从而获得带有烯烃的产物。该策略成功地实现芳基碘化物C(sp2)-I键断裂,从而获得关键的芳基Pd(I)-自由基中间体A。值得注意的是,这些新方法依赖于使用有机卤化物(碘化物、溴化物等),这限制了该方法的合成适用性。如果能够使用芳基亲电试剂代替,则能够大大扩展底物的适用范围。本文中,作者报道了在温和可见光诱导的钯催化条件下,实现芳基三氟甲磺酸盐(三氟甲磺酸酯)1的C(sp2)-O键的断裂,从而形成芳基Pd(I)-自由基A1,再经连续的1,5-HAT(A1→B1)/分子内环化/再芳构化步骤后,获得具有价值的羟吲哚产物2(Scheme 1b)。
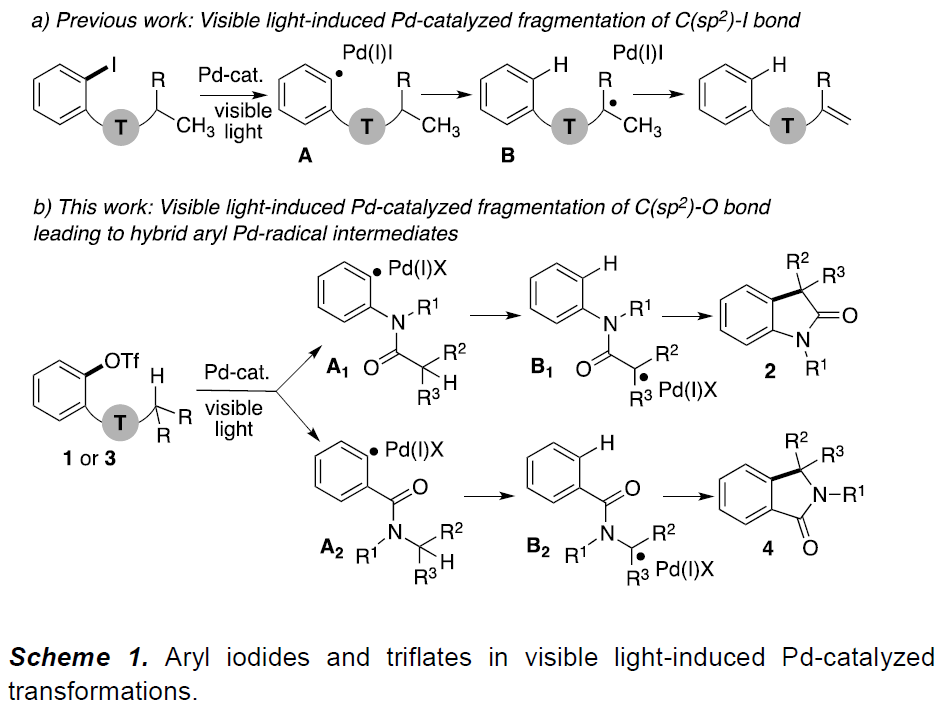
受芳基卤化物成功断裂形成Pd(I)-自由基中间体的鼓舞,以及Li课题组在紫外线诱导下实现C(sp2)-O键的断裂(eq. 1)的启发,作者开始研究是否可通过可见光/钯催化条件,实现芳基三氟甲磺酸酯生成芳基Pd(I)-自由基中间体的过程。

作者选择了芳基三氟甲磺酸酯1作为分子内偶联的底物。此外,Hartwig文献报道,酰胺分子内α-芳基化是构建羟吲哚骨架的有效方法,尽管该方法具有高收率和对映选择性,但依赖于使用强碱,从而限制了底物的范围。
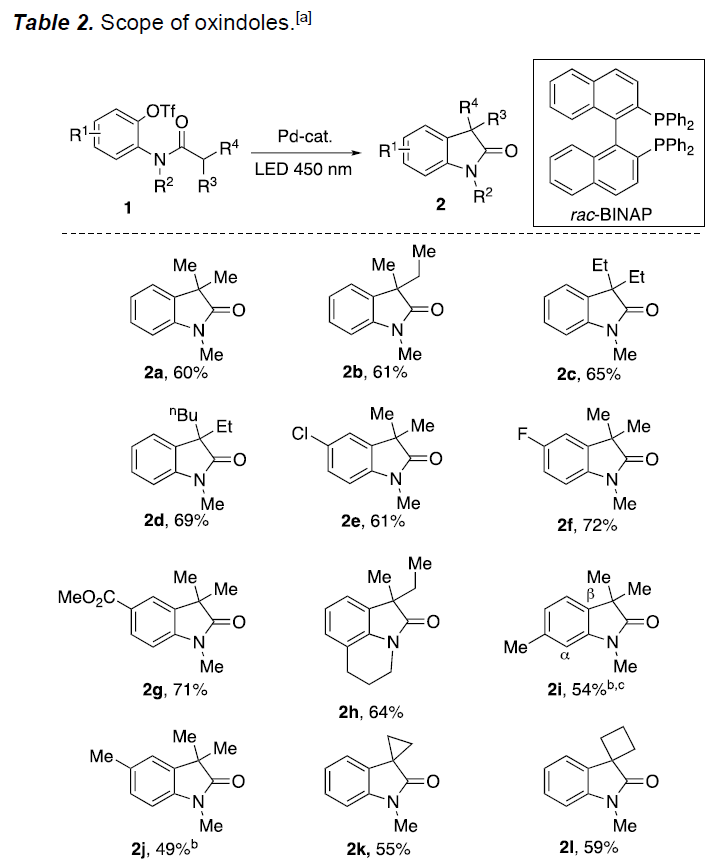
首先,作者以三氟甲磺酸酯1a作为模型底物,进行分子内α-芳基化反应的研究(Table 1)。作者使用以前优化的反应条件用于此反应,但并未获得目标产物,然而当使用双[(2-二苯基膦基)苯基]醚(Dpephos)作为配体,在DMF溶剂中,可获得低收率的吲哚产物2a(entries 1-2)。进一步的优化表明,添加NaI可使分子内芳基化反应进行得更好(entry 3)。大量的配体筛选表明, 2,2′-双(二苯基膦基)-1,1′-联萘胺(rac-BINAP)作为最佳配体,可使羟吲哚2a的收率提高至41%(entry 4)。同时,作者也对影响Pd催化的添加剂进行了筛选,但并未取得好的结果(entries 5-6)。溶剂的优化表明,该反应在1,4-二氧六环中可获得62%收率的2a。对照实验表明,催化剂、光和添加剂都对反应至关重要(entries 8-9)。此外,使用自由捕获剂2,2,6,6-四甲基哌啶1-氧基(TEMPO)时,导致收率大大降低,从而表明该反应可能涉及自由基中间体(entry 10)。

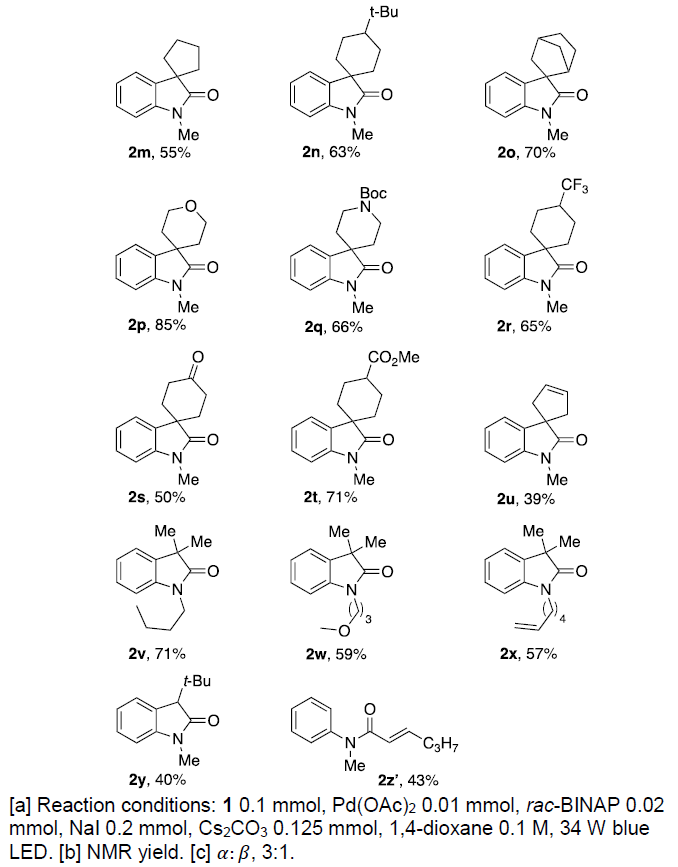
在获得上述最佳反应条件后,作者开始对底物1进行了扩展(Table 2)。首先,使用三氟甲磺酸酯1a可获得60%收率的羟吲哚2a,同时可在α-碳旁引入各种烷基(乙基、叔丁基等)获得相应的羟吲哚产物2b–2d,而芳环上具有卤素和酯官能团的底物均以高收率获得产物2e–2g。同样,以64%产率获得三环羟吲哚产物2h。芳基环上的烷基取代基也与体系兼容,可进一步扩展到螺环产物的合成,如螺环丙基-(2k)、环丁基-(2l)、环戊基-(2m)和环己基-(2n)羟吲哚等。降冰片烷衍生物1o也能够平稳进行反应获得2o(收率70%),杂原子的引入同样获得相应的羟吲哚产物2p–2r。值得注意的是,在标准条件下,对碱敏感的官能团(如酮和酯)具有良好的耐受性,可分别以50%和71%的产率获得羟吲哚产物2s和2t(传统的Pd催化方法不能获得)。环戊烯衍生物1u也可转化成相应的羟吲哚2u,但收率中等。在N原子处取代的改变分别获得71%、59%和57%的羟吲哚2v–2x。值得注意的是,该转化能够获得3-单取代的羟吲哚2y,尽管产率为40%。但是,用空间上要求较少的正戊基部分取代大体积的叔丁基时,导致形成无环的不饱和酰胺2z’(主要产物)。
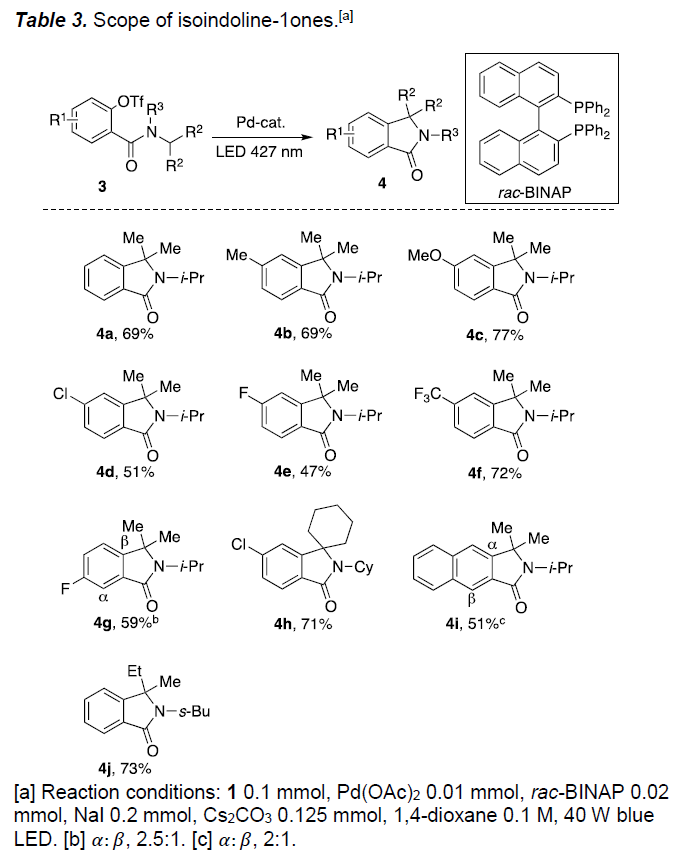
值得注意的是,上述合成羟吲哚2的方法是在富电子的芳香环上进行环化(Scheme 1b)。作者假设在电子相反的情况下(A2→B2)可以进行类似的环化反应,从而获得具有价值的异吲哚啉-1-酮衍生物4。因此,在优化的反应条件下研究了苯甲酰胺3a的环化反应(Table 3)。当以异丁二烯-1-酮3a作为底物时,可获得69%收率的4a。同时,具有烷基、甲氧基、卤代、三氟甲基和萘基的不同取代的苯甲酰胺(3b–3g,3i)均以中等至良好的产率获得相应的异吲哚啉-1-酮衍生物。此外,环己基和异丁基酰胺底物也能够进行相应的的环化,分别获得71%和73%收率的4h和4j。
为了进一步了解分子内C-H烷基化反应的机理,作者进行了一些氘代实验。当使用TEMPO时(Table 1, entry 10),导致收率急剧下降,同时后续的氘代实验(eq. 2),进一步证明了反应涉及自由基参与。首先,作者研究了氘代三氟甲磺酸盐1a-D的分子内芳基化作用,1a-D经C-O键断裂和1,5-HAT生成B1–D中间体,在没有电子效应影响的情况下,芳环的C2或C6位置均具有相同的环化可能性,从而导致羟吲哚2a-D,氘标记保留约47%。实际上,使用三氟甲磺酸酯1a-D的实验完全支持了上述预测,从而导致在芳香环上含有48%氘的羟吲哚2a-D。此外,使用D-标记的三氟甲磺酸酯1a’-D进行环化,产生了单一产物羟吲哚2a-D,与先前的实验一样,在氘标记掺入的预测水平和观察水平之间观察到极好的一致性。这些氘标记实验排除了潜在的直接C-H活化机制(1a’-D→i→2a-D),由于观察到D标记的C2位置的完全保留(eq. 3)。另外,通过三氟甲磺酸酯1aa产生开环的二烯酰胺2aa(eq. 4)的自由基钟实验以及DMPO的自旋捕捉实验进一步证实了自由基反应的机理。

根据上述的实验和相关文献的查阅,作者提出了一种可能的反应机理,涉及关键芳基Pd-自由基中间体A1的形成(Scheme 2)。首先,底物1a在钯和可见光催化下获得关键中间体A1,经1,5-HAT形成杂烷基自由基B1,紧接着在芳环C1上进行分子内环化形成和重新芳构化后,即可获得目标羟吲哚产物2a。目前,尚不清楚C-O键断裂步骤的具体细节以及NaI的确切作用。此外,作者也对关键芳基Pd-自由基中间体A1的形成提出了几种可能的路径。在path A中, 1a与活性光激发的Pd络合物之间经SET形成三氟甲酸酯芳基自由基阴离子,再经C-O键断裂,则生成A1。在path B中, 1a与Pd(0)经氧化加成形成Pd(II)中间体D1,再经可见光诱导的C-Pd键均化,从而形成A1。在path C中,D1和NaI之间发生配体交获得更稳定的芳基-Pd(II)-I中间体E1,再经可见光诱导的C-Pd键均化可将E1转换为A1。在path D中,中间体E1经还原消除生成芳基碘化物5,再与光激发的Pd催化剂经SET,从而生成A1。值得一提的是,一些机理实验,包括对底物模型的EPR研究、化学计量反应速率比较、早期阶段监控平行反应等,表明了上述四种方案的可行性。然而,需要更详细的力学研究才能建立起这种新颖转化的精确机制。

总结
伊利诺伊大学的Vladimir Gevorgyan教授团队报道了将可见光与钯催化相结合,实现芳基三氟甲磺酸酯中C(sp2)-O键断裂,从而获得芳基Pd(I)-自由基中间体,随后再经1,5-HAT/环化/芳香化获得不同取代的羟吲哚和异吲哚啉-1-酮衍生物。此外,该方案可以合成传统的Pd催化体系不易合成的具有碱性敏感基团(如酮和酯)的羟吲哚产物。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!














No comments yet.