
作者:石油醚
引言
近日,Relay Therapeutics、Acceledev Chemical、苏利制药和南京药石等公司科学家合作阐述了化合物 2 合成路线的战略演变,从初始的外消旋合成发展至多种生物催化策略,最终建立了一条基于水解动力学拆分(HKR)的高度可放大不对称工艺。本工作研究表明,整合不对称催化、杂质管理与工艺强化,可将复杂合成转化为可放大的实用工艺,有力支持早期临床开发。
From Chiral Separation to Asymmetric Synthesis: Route Scouting and Early Process Development of a Chiral Tetrahydrofuran−Pyrazolamine Intermediate of CDK2 Inhibitor RLY-2139
Seema Bag, Andre Lescarbeau, Allen Qinglin Che, Vishnu Karnati, Zhongbo Fei, Liyang Wang,
Dehai Zhang, Jun Liu, Fan Jiang, Hai Wang, Gang Huang, Pengfei Shen, Jing Wang, Mingying Zhu,
Yonghui Lu, Fanfan Meng, Baldip Kang, Surendra Singh,* and David Tschaen*
Org. Process Res. Dev. 2026. DOI : 10.1021/acs.oprd.5c00354
正文
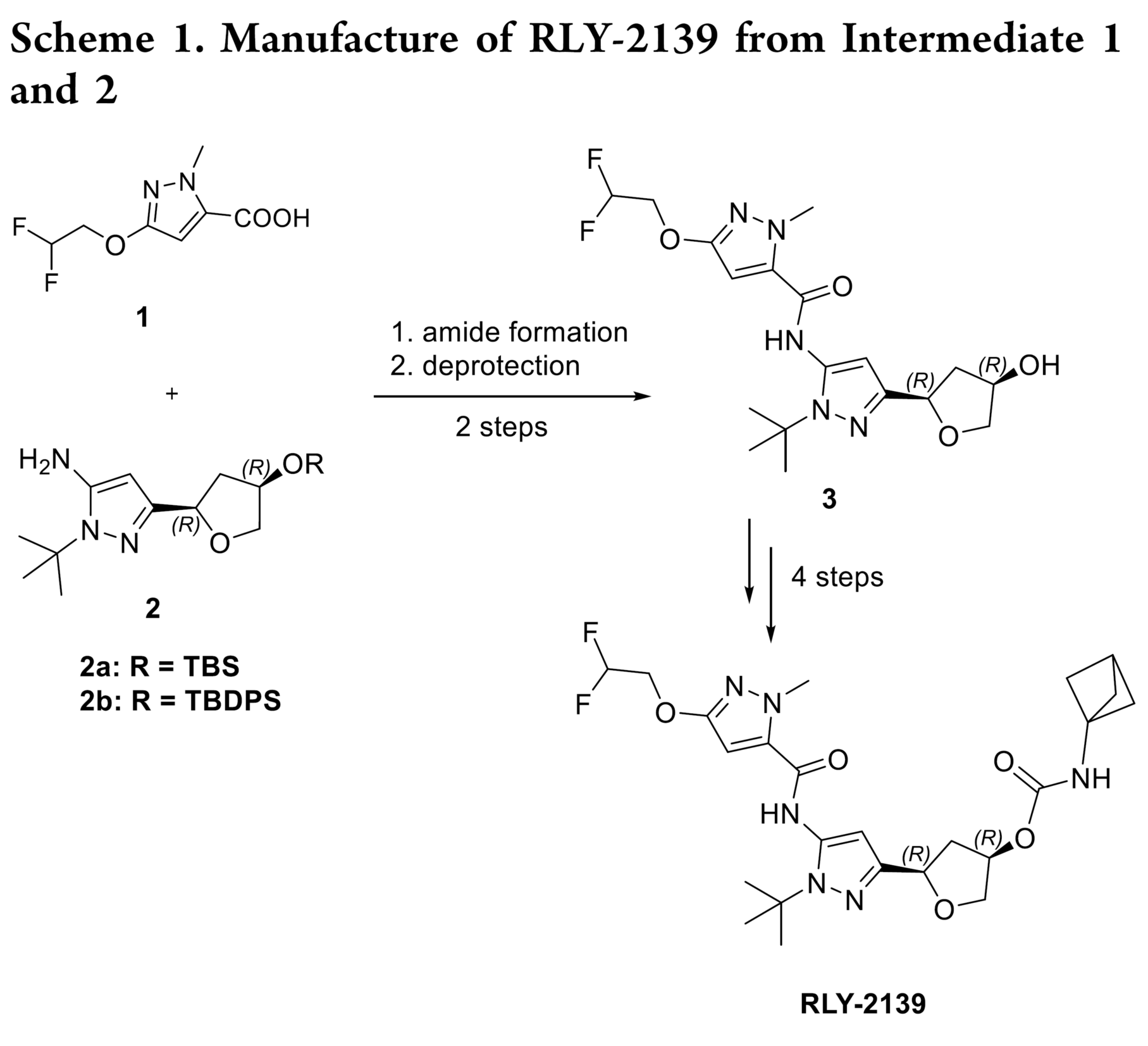
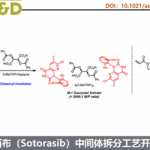
RLY-2139 是 Relay Therapeutics 开发的一种用于治疗ER+/HER2− 乳腺癌的高效选择性 CDK2 正构抑制剂。其通过选择性抑制 CDK2/细胞周期蛋白 E 复合物发挥作用,且该复合物是 CDK4/6 抑制剂耐药肿瘤中驱动细胞周期的关键因子。RLY-2139的合成采用了一种汇聚式路线,即通过两个高级中间体酰胺缩合,并进行后期官能团化(Scheme 1)。其中,化合物 2 的构建是RLY-2139合成中的最大难题。
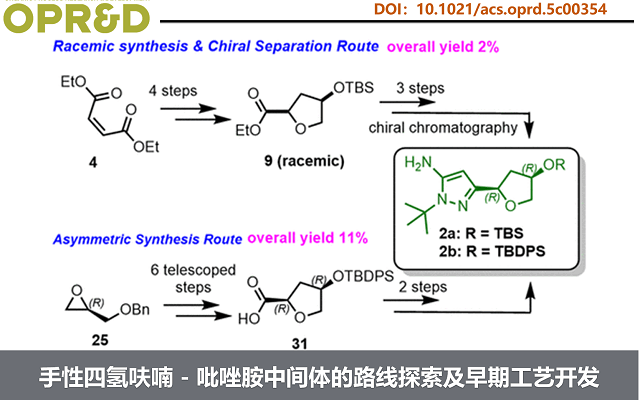
早期,化学家经7步合成(Rac)-2a,手性分离获得2a。虽然该路线可支持早期临床前供应(Scheme 2),但因整体产率低、依赖制备型色谱,且需丢弃无效对映体导致物料利用率低,难以满足cGMP生产和大规模临床制造的需求,亟需一条更高效、更具对映选择性的新路线。
基于此,Relay Therapeutics、Acceledev Chemical、苏利制药和南京药石等公司科学家合作阐述了化合物 2 合成路线的战略演变,从初始的外消旋合成发展至多种生物催化策略,最终建立了一条基于水解动力学拆分(HKR)的高度可放大不对称工艺。开发过程中重点关注对映体纯度控制、杂质管理、步骤合并、避免低温操作及放大稳健性等关键工艺要素。该研究凸显了逆合成灵活性与战略化分子设计在复杂药物中间体工艺开发中的核心作用。
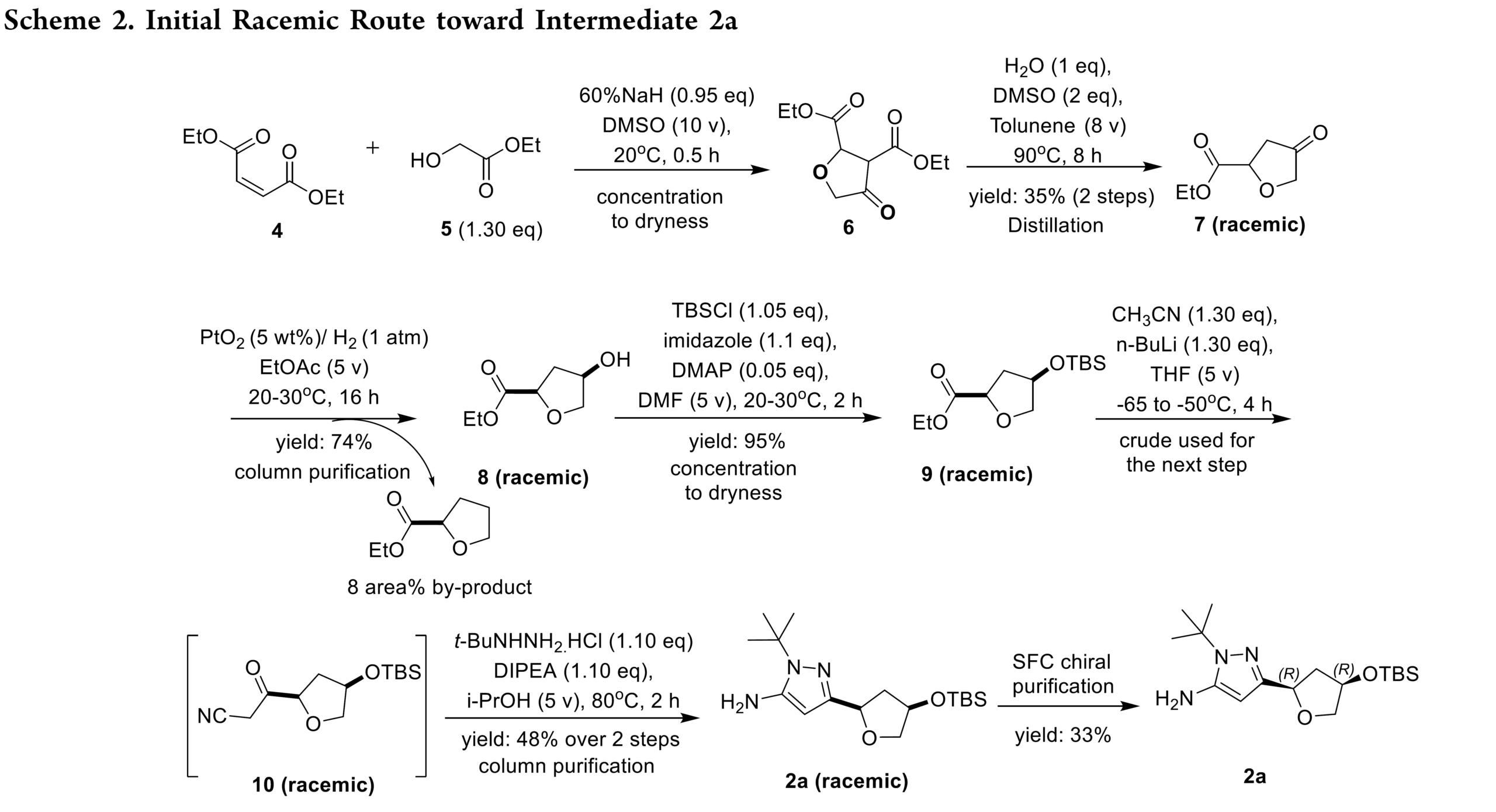
首先, 化合物 2a 的早期合成路线(Scheme 2)以马来酸二乙酯 4 和 5 为起始原料,经 Michael 加成和 Dieckmann 缩合得到纯度 >97%(GC)的 Rac–7。Rac-7 经催化氢化高选择性生成目标产物 cis–8,但伴随约 8%(面积)的过度还原杂质,需色谱纯化去除。该步骤及后续 TBS 保护后浓缩析出 Rac–9 均造成物料损失。Rac–9 经 n-BuLi 介导的氰甲基化与叔丁基肼环合,再次依赖色谱纯化获得 Rac–2a。最终通过手性色谱分离以 33% 收率得到目标对映体 2a。多个低收率步骤与重复纯化导致总收率仅为 2%。尽管该路线满足了初期 7 kg 早期开发供应,但因效率低、物料通量差且严重依赖色谱,难以放大。上述局限推动了更可放大、可持续且适用于 cGMP 生产的替代工艺的系统性开发。
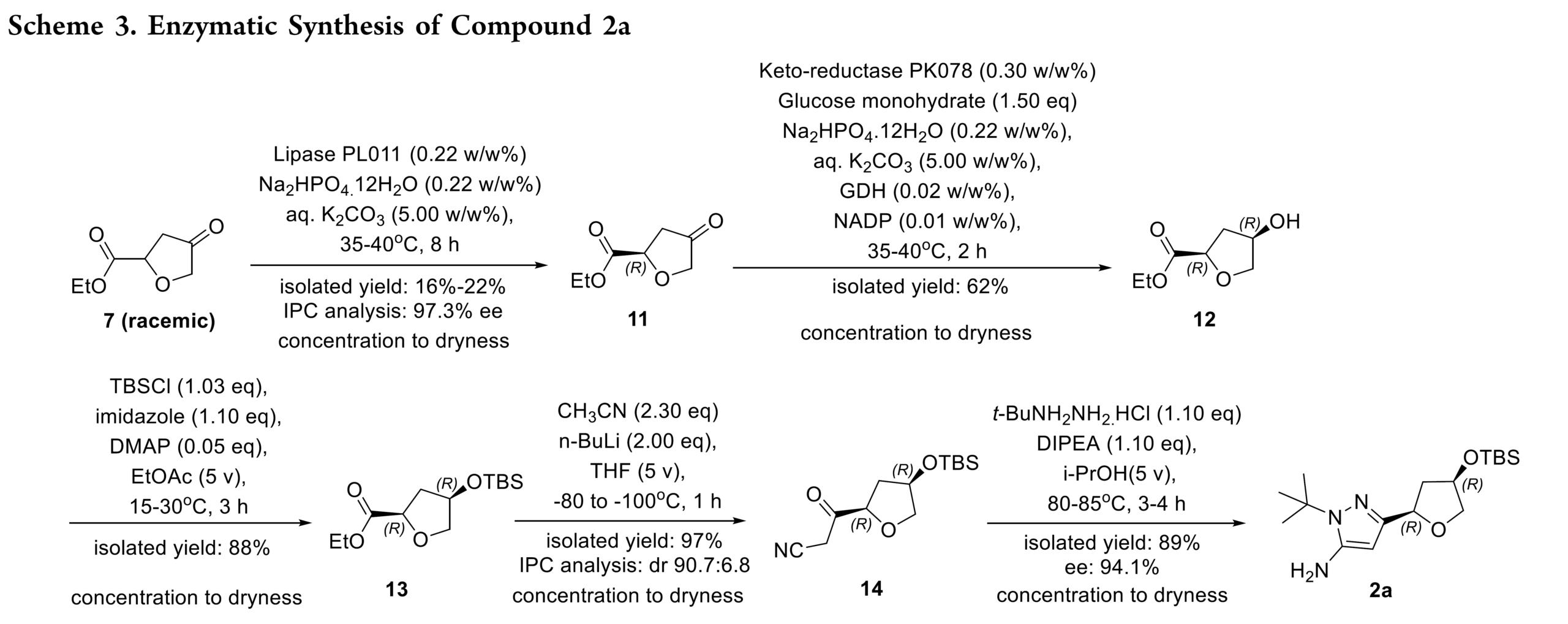
其次,作者希望利用生物催化来提高上述合成路线的效率(Scheme 3)。即以Rac–7作为原料,经脂肪酶进行动力学拆分和酶促还原构建第二个手性中心来提高选择性简化合成流程。基于此,筛选发现,脂肪酶可实现Rac–7的拆分获得所需对映体 11(97.3% ee),但实验室规模分离收率仅为 16–22%。11经酶促还原建立第二立体中心,TBS 保护得手性中间体 13;再经氰甲基化及与叔丁基肼环合,以 89% 收率得到化合物 2a。然而,深入分析发现初始脂肪酶步骤存在关键缺陷:S-酯完全水解,但目标 R-酯也发生部分水解,导致总收率降低。另外一种策略是:先用酮还原酶(KRED)还原酮基,再进行脂肪酶介导的酯水解。部分 KRED 可实现完全转化且专一生成顺式产物,但后续脂肪酶无法选择性水解非目标 (S,S)-对映体,导致分离困难、纯化效率低,收率仍不理想。
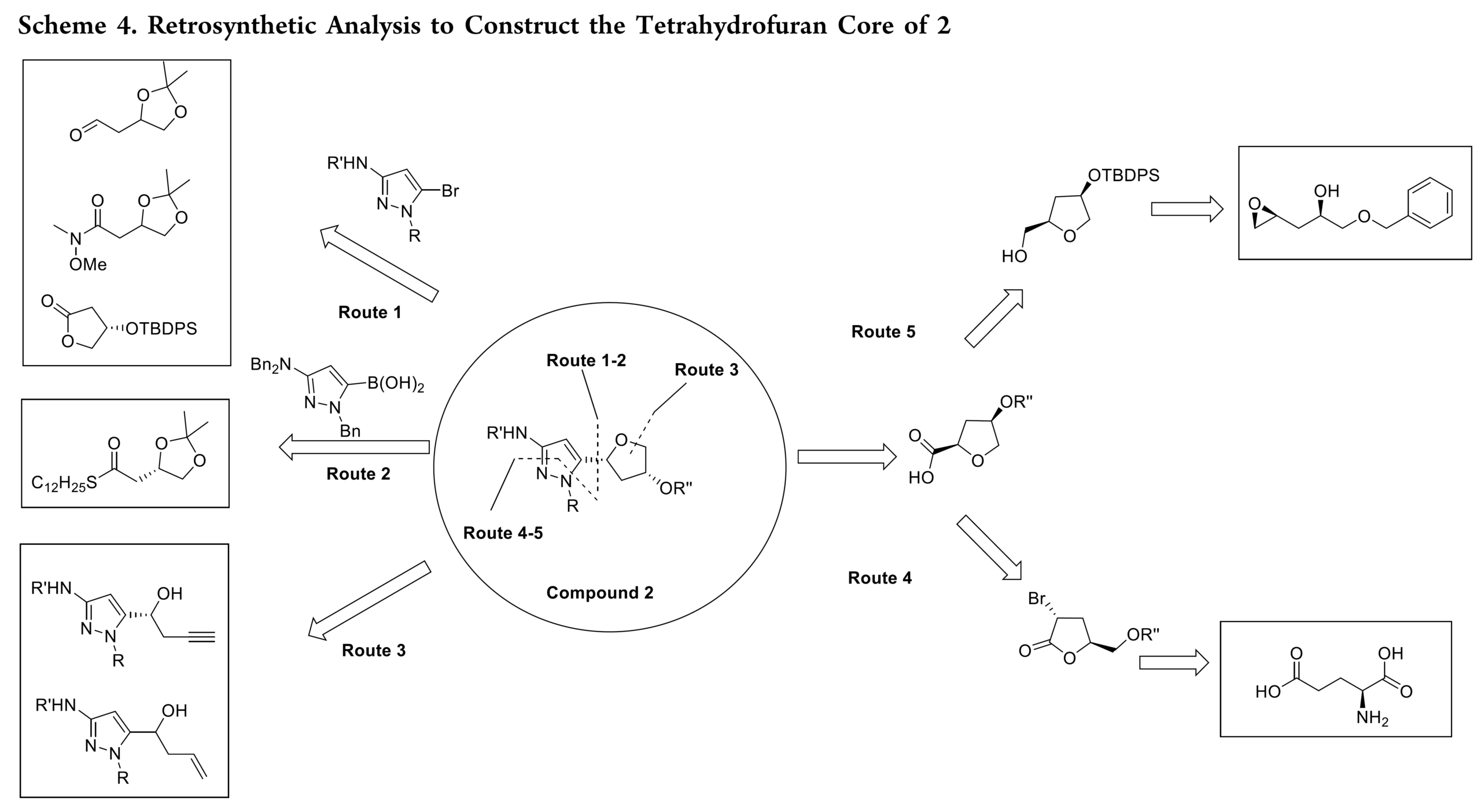
由于酶促动力学拆分路径存在严重的问题,作者对2进行了逆合成分析,并确定了五条路线,如Scheme 4所示。即初步分析筛选出五条不同路线,各具独特战略思路:路线 1 和 2 聚焦于通过 C−C 键偶联吡唑单元与预功能化的四氢呋喃片段;路线 3 探索以后期 四氢呋喃环闭合作为关键成键步骤;路线 4 和 5 则采用手性池策略,分别利用两类市售手性原料构建 四氢呋喃环,再通过初始外消旋合成中验证有效的稳健化学方法,汇聚式引入吡唑基团。
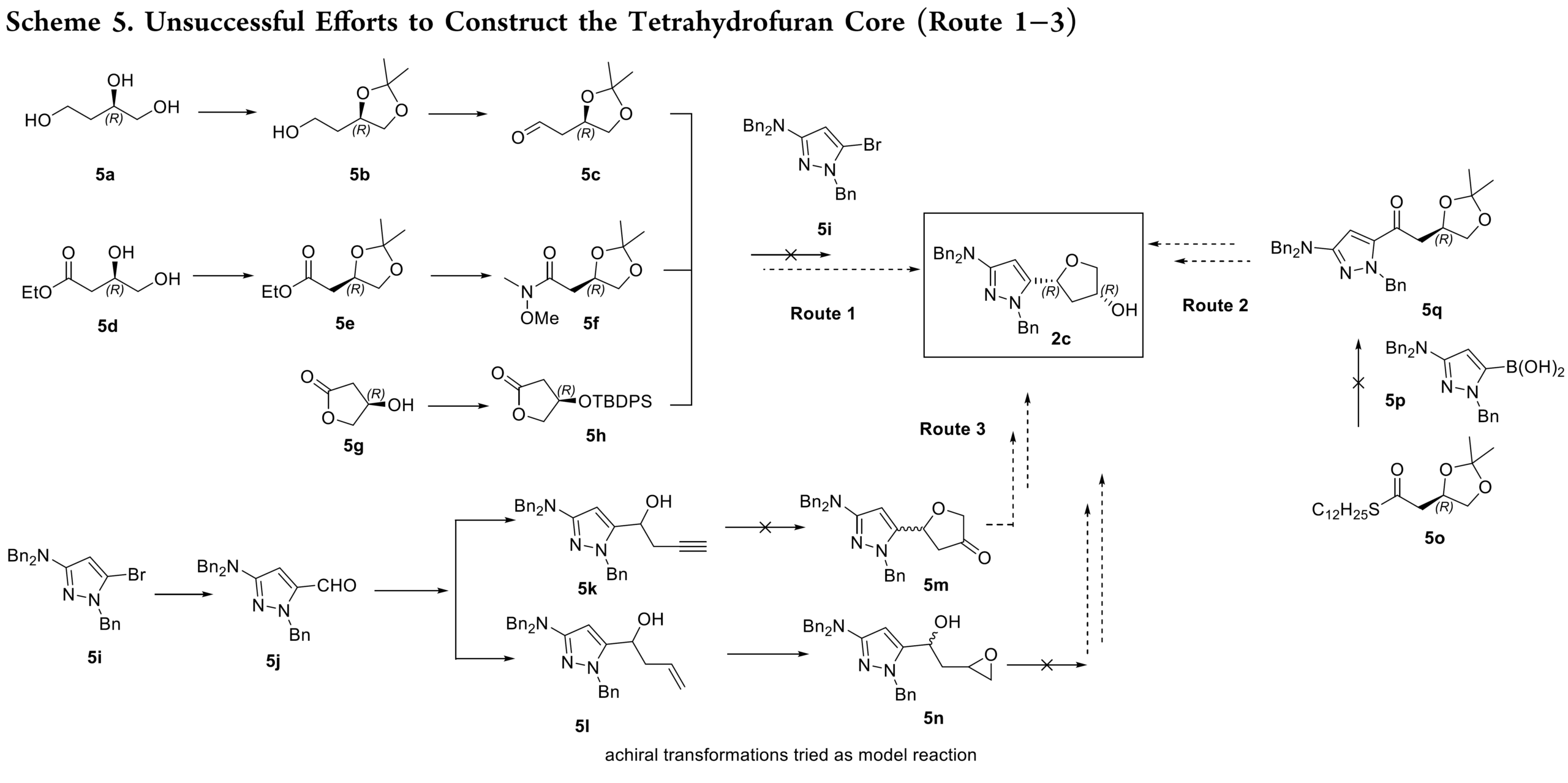
在逆合成分析提出的五条路线中,路线 1–3 率先被研究,但因存在重大技术难题,均被放弃,无法用于工艺开发(Scheme 5)。其中:路线1中吡唑衍生物与亲电性四氢呋喃醛( 5c、Weinreb 酰胺 5f 和内酯 5h)片段无法偶联;路线2中5o和5p 的Liebeskind−Srogl 反应难以发生;路线3中采用分子内环化策略难以构建四氢呋喃环。
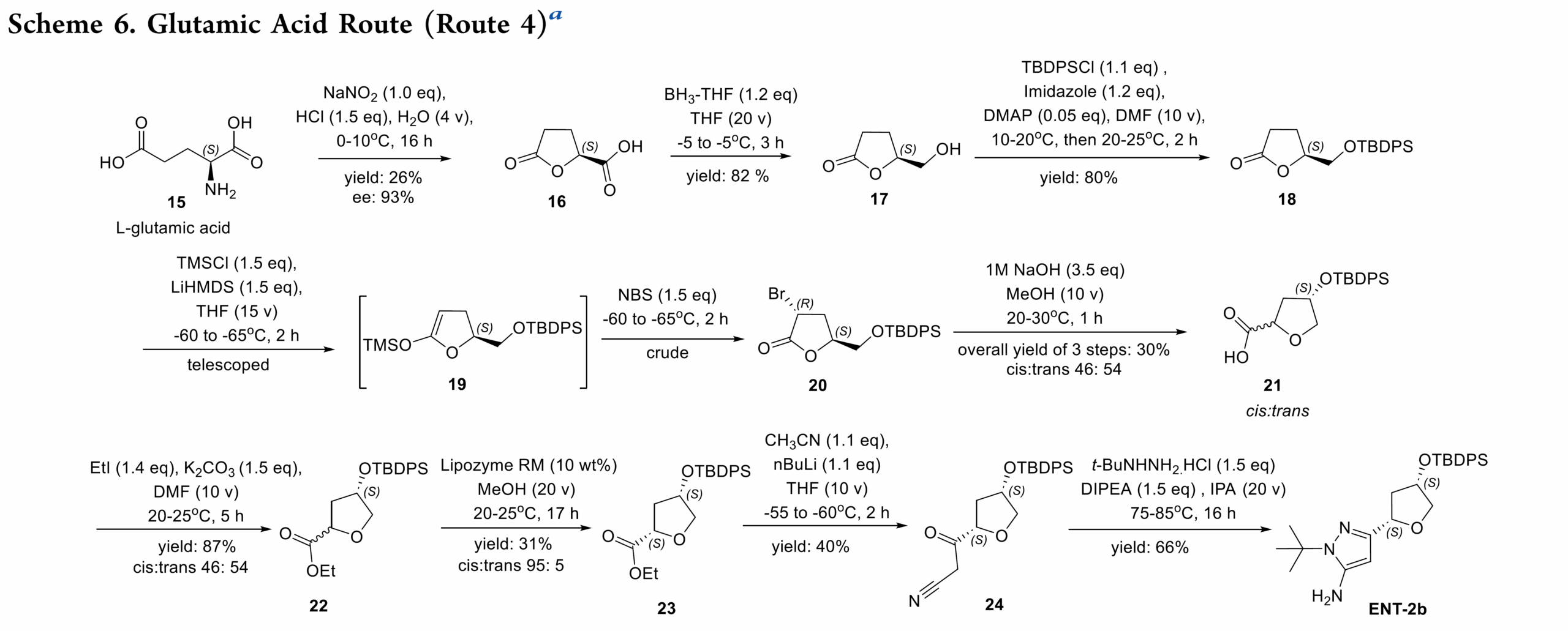
路线4:路线 4首次成功构建了目标四氢呋喃核心,验证了不对称合成的可行性。然而,该路线因手性消旋、产率低的步骤以及复杂的中间体处理在内的诸多难题,而不适用于放大生产。即以L-谷氨酸 15作为原料,经重氮化反应内酯 16(93% ee, 26% yield)且存在明显手性流失。特别,16 因高水溶性和缺乏紫外吸收基团,难以分离与表征,不利于工业化。16经还原和 TBDPS 保护得中间体 18;再经 LiHMDS 脱质子化、TMS 烯醇醚化及溴化,得到约 9:1 的非对映体混合物 20。随后发生碱催化的开环与 TBDPS 迁移,再环合生成顺反比为 46%:54% 的 22 混合物。22采用 Liposyme RM 酶促拆分,选择性水解tarns-异构体,得到顺式占优的 23(cis:trans = 95%:5%)。碱性后处理去除水解产物后,分离出 23。最后经氰甲基化及叔丁基肼环合,得到最终产物 ENT-2b(S,S-对映体)。
路线5:克服先前路线的局限性(Scheme 7),通过在整个合成过程中保持手性完整性,最终得到 2b。即以苄基(R)-(-)-缩水甘油醚25作为原料,经过乙烯基氯化镁的格式加成、m-CPBA 介导的末端烯烃环氧化反应、Jacobsen 水解动力学拆分(HKR)、甲硅烷基化和重排、氧化和氰甲基化、构建吡唑形成反应得到 2b。2b经酰胺偶联得到中间体 33,再经叔丁基二苯基硅烷保护基脱保护,得到 3(Scheme 8)。工艺优化表明,m-CPBA 和 Jacobsen 催化剂的质量对反应性能有关键影响。目前,该优化路线成功放大了两批,共得到 28 kg的 3。总体而言,与原路线相比,该工艺流程得到简化,生产成本降低约 50%。
2b合成关键工艺路线优化如下。
- Step 1:格式加成[181 kg 26]。25与乙烯基溴化镁进行格氏反应时,因溴离子引发环氧开环而产生溴化物杂质。通过改用0 equiv的乙烯基氯化镁得以解决。其中,过量的乙烯基格氏试剂增强其对环氧的加成竞争力,抑制了氯离子开环副反应。优化后,可在85kg的环氧作为原料,在−5 °C下缓慢、受控地滴加乙烯基氯化镁,历时12.5小时,有效控制温度,以88%的纯度收率得到26,副产物氯代开环物含量仅约6%。该杂质在后续工序中被彻底清除。
- Step 2:环氧化[184 kg 27]。环氧化采用过氧化氢与有机催化剂体系,因显著放热和气体释放存在安全风险。因此改用 m-CPBA 作为氧化剂,并通过DSC分析表明mCPBA 体系的反应能量显著低于 H₂O₂ 体系。商用 m-CPBA 含有过氧化苯甲酰,会抑制后续 Jacobsen HKR 反应。通过引入三乙胺处理粗产物,有效分解该杂质,保障后续步骤的高效进行。
- Step 3:HKR[103.4 kg 28]。工艺过程中的关键步骤。初始反应速率慢主要源于三个因素:前步残留的苯甲酰过氧化物杂质、Jacobsen 催化剂活性不足,以及批次间性能波动。苯甲酰过氧化物在27后处理中通过三乙胺洗涤去除;催化剂性能通过使用前小试进行筛选以确保达标;批次粘度因三醇副产物积累而升高,通过增加溶剂体积得以降低。残余三醇在中间体 31 成胺盐析出时被彻底清除。
- Step 4/5:硅烷化与重排[107 kg 29/223 kg 30]。硅烷化在−10 °C进行,有效抑制氯离子介导的环氧开环,所得中间体29中该杂质低于3%。该中间体直接用于后续反应。随后的路易斯酸催化重排(使用BCl₃或Et₂AlCl)高效完成。
- Step 6:氧化[84 kg 31]。最初采用丙酮中的催化 TEMPO-TCCA 体系,延长加料时间会导致产率显著下降,因 TCCA 在丙酮中不稳定,难以满足大规模生产。改用乙腈作为溶剂后,TCCA 稳定性提高,问题得以解决。产物以 S-(−)-1-苯乙胺成盐析出,实现首次分离,并作为高度串联路线中的关键质量控制点。所得固体经过滤、MTBE 洗涤后转化为游离碱(纯度 4% AUC,cis:trans = 96.6:3.1,KF = 0.22%),直接用于下一步。
- Step 7:酰化[82.4 kg 31]。首先用CDI将酸31活化为酰基咪唑,随后加入经格氏碱处理的氰乙酸悬浮液。使用iPrMgCl作碱时产生大量杂质,经与标准品比对确认为酸31的异丙酯。改用位阻更大的t-BuMgCl后,相应叔丁酯杂质显著减少,控制在8%(面积)。残留杂质可能源于市售试剂中的微量叔丁醇或格氏试剂因空气暴露发生的氧化。工艺放大至中试规模后,杂质进一步降低,归因于更高纯度的原料和更有效的氮气保护。
- Step:缩合[26 kg 2b]。最终的环合步骤形成了关键的吡唑环。这里的主要挑战在于其中一个手性中心的差向异构化。我们的优化研究发现,差向异构化与延长反应时间和增加二异丙基乙胺(DIPEA)的当量数直接相关。最佳条件是在 80°C 下使用 5 当量的 DIPEA,这样反应可在 16 小时内完成。这种谨慎的控制将差向异构化率降至约 5.9%,这一杂质水平在后续步骤中得以成功去除。尝试将最终产物 2b 制成固体盐均未成功,因此该化合物以油状物的形式进入合成的下一阶段。
2b转化为中间体3的工艺优化
化合物 2b 经两步反应转化为中间体 3,过程与方案 1 中的初始化学反应类似。
酰胺缩合。化合物 2b 在 CMPI 作用下发生酰胺偶联生成产物 33。通过重结晶,仅需一步纯化即可去除前步残留的差向异构体及其他工艺杂质,显著提升产物纯度,最终以纯净结晶固体形式分离出 33。
脱保护。TBDPS 脱保护生成目标分子 3 的最后一步起初因产物物理性质不佳而困难。粗产物为粘稠固体,难以过滤。问题主要源于两方面,经工艺优化后得以解决。首先,中间体 33 经彻底重结晶并进行活性碳吸附处理,确保起始物料高纯度,对获得洁净终产品至关重要。其次,残留 TBAF 是导致粘性的主因。中性或碱性水洗无效,改用稀盐酸水溶液洗涤可高效去除。最后,增加一次活性碳媳妇处理以消除重结晶后的灰色,得到纯净白色固体。上述纯化策略共同保障了终产品的高质量,满足后续开发要求。
结论
本研究开发了药物RLY-213所需手性四氢呋喃吡唑胺中间体化合物 2 的合成工艺。酶促法和手性池法虽理论可行,却因放大困难和严重手性消旋,不适合大规模生产。经过系统筛选最终确立了一条以手性环氧化物 25 为起始原料、结合 Jacobsen 水解动力学拆分(HKR)的稳健可放大路线。优化后的八步合成有效解决了多步合成的关键挑战,主要体现在以下方面:
- 收率提升:通过阶段适配的工艺优化,不对称路线总收率达 11%,显著优于原路线的 2%;
- 安全与效率提升:采用安全的环氧化条件,并实现硅基保护与重排的串联操作;
- 杂质控制有效:设计了多种杂质清除策略,包括 HKR 步骤的新颖后处理方法;
- 工艺强化:全程无需色谱纯化,实现物料连续高效流转。
该路线立体选择性高,已成功放大至生产 28 kg 以上高级中间体 3,具备未来 cGMP 生产的可行性。本工作研究表明,整合不对称催化、杂质管理与工艺强化,可将复杂合成转化为可放大的实用工艺,有力支持早期临床开发。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/


















No comments yet.