
本文作者:杉杉
摘要
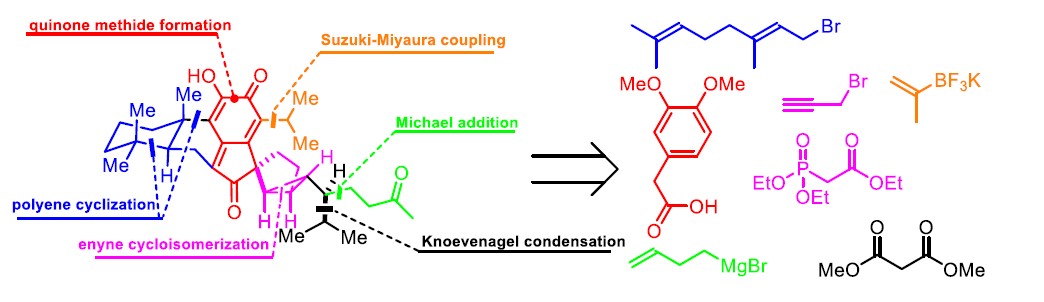
近日,香港中文大学彭小水课题组在Angew. Chem. Int. Ed.上发表论文,首次报道了天然产物cryptotrione(1)的全合成路线,涉及三个关键步骤,即氯化铂(Ⅳ)催化烯炔环异构化构建双环[3.1.0]己烯骨架,路易斯酸引发多烯环化构建松香烷(Abietane)型三环二萜骨架,在侧链非对映共轭加成实现叔碳中心的控制。
前言
自1983年从日本柳杉家族中分离出第一个cryptotrione成员以来,已有九个家族成员被分离,包括chamaecydin(2),isochomaecydin(3),cryptoquinonemethides D和E(4和5)。在2010年,郭悦雄小组成功于日本柳杉(Cryptomeria japonica)的树皮中首次分离出一种新型的C35-萜烯,即Cryptotrione 1(Figure 1)。作为结构最为复杂的Cryptotrione,它具有抗口腔鳞癌(human oral epidermoid carcinoma,KB)细胞的生物活性,IC50值约为6.44±2.23μM,仅比临床使用的抗癌药依托泊苷(etoposide)稍弱(IC50为~2.0 μM)。尽管史一安课题组已报道了双环[3.1.0]己烯核骨架的构建,但尚未报道其全合成路线。在此,香港中文大学彭小水课题组通过铂催化实现构建双环[3.1.0]己烯结构后,再与亚甲基醌化合物反应,从而实现了cryptotrione(1)的全合成。论文发表在Angew. Chem. Int. Ed.上。
Total Synthesis of Cryptotrione
M.-Y. Lyu, Z. Zhong, V. K. Y. Lo, H. N. C. Wong,* X.-S. Peng*
Angew. Chem. Int. Ed. ASAP DOI: 10.1002/anie.202009255
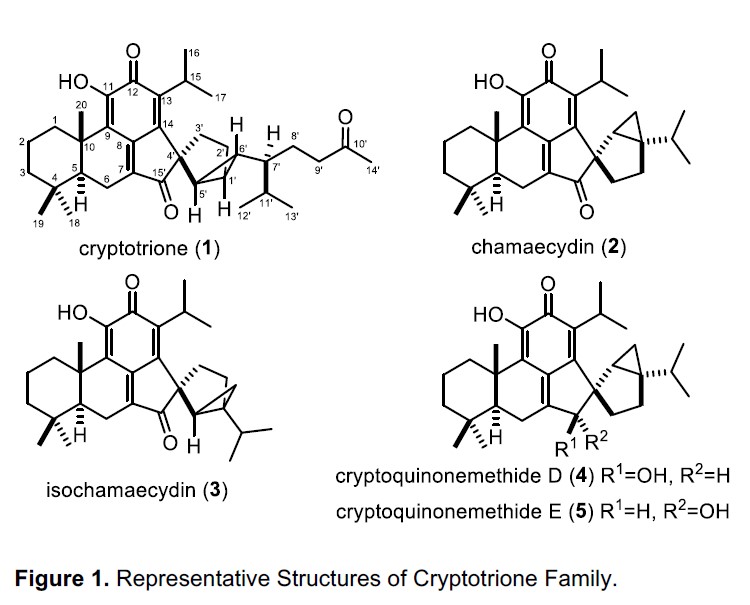
Figure 1 Cryptotrione 家族结构
路线设计
逆合成分析发现,cryptotrione(1)可由双环[3.1.0]己烷单元和亚甲基醌化合物组成(Figure 2)。基于金或铂催化1,n-炔烃环异构化构建双环[n.1.0]己烷衍生物的文献总结,作者认为可将1,5-烯炔化合物10通过环异构化合成关键的双环[3.1.0]己烷中间体9。此外,通过1,5-烯炔底物的官能团修饰,也可有效合成其他cryptotrione家族成员。以市售高藜芦酸11为起始物料,通过已知方法即可合成1,5-烯炔前体10。随后,醇9经烷基化生成多烯8。8通过路易斯酸引发的立体选择性发生环化和异丙基化反应,生成反式多环化合物7。紧接着,引入侧链得到烯烃6。最后,形成的亚甲基醌化合物再进行Wacker氧化,即可合成cryptotrione(1)。
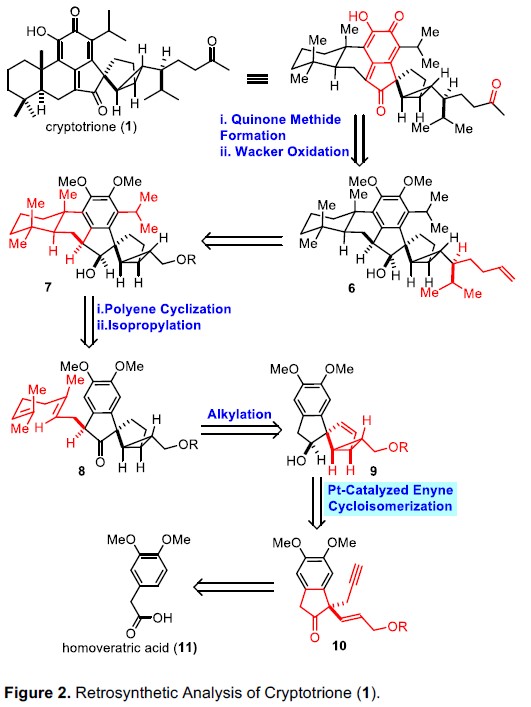
Figure 2 Cryptotrione的逆合成分析
双环[3.1.0]己烷骨架13的构建(Scheme 1)
以市售高藜芦酸(11)作为初始底物,经碳烯插入法可制备各种烯炔12a–12e,继而转化为所需的双环[3.1.0]己烷化合物13a–13e,而不是13´a–13´e。同时,为了了解烯炔12a–12e中保护性羟基的立体效应,作者通过非对映选择性还原制备了C15′-α-甲硅烷氧基-1,5-烯化合物和C15′-β-甲硅烷氧基-1,5-烯化合物。在Toste’s的Au催化条件,C15′-α-甲硅烷氧基-1,5-烯炔会生成不希望的双环[3.1.0] 己烯化合物(与cryptotrione(1)相对构型不同)。因此,作者将注意力转向C15′-β-甲硅烷氧基-1,5-烯炔12a–12e的环异构化,从而形成所需的立体化学的关键前体13a–13e。
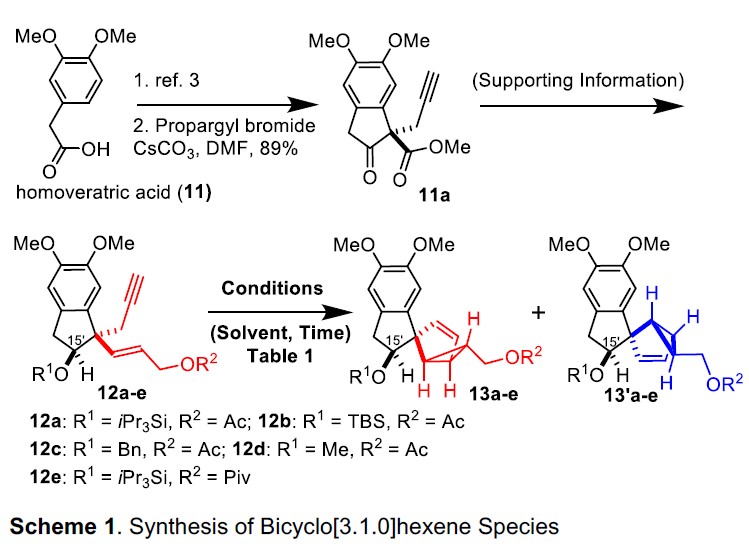
当使用10%Ph3PAuCl/SbF6催化剂时,烯炔12a完全分解(Table 1, entry 1),但在100℃的甲苯溶剂中,使用20mol%的PtCl2时可顺利将12a的烯炔异构化,获得44%收率和dr>30:1的13a(entry 2)。在相同条件下,含有较小保护基的12b(TBS与iPr3Si)收率略低(entry 3)。具有β-苄氧基的烯炔12c也以30%的收率得到期望的13c。相反,由于空间位阻不足,带有甲氧基的烯炔12d结果较差(entry 6)。将R2上的乙酰基保护基替换为新戊酰基,收率略有提高(entries 4-5),这表明C15’处的β-三异丙基甲硅烷基具有立体选择性。然后,当以10 mol%的PtCl4作为催化剂时,可将收率提高至75%(entries 7-8)。此外,使用10 mol%的PtCl4催化剂在115℃下,该异构化反应放大至15g时,以71%的收率得到13e(entry 8)。
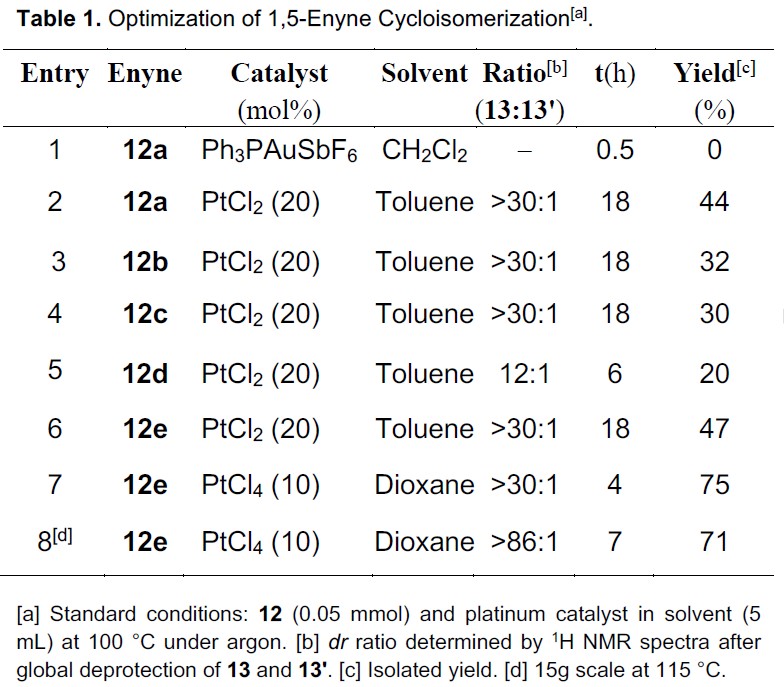
二醇骨架21的构建(Scheme 2)
在获得关键的中间体13e后,作者开始对二醇21进行了合成。13e经脱甲硅烷基化和氢化后,获得75%收率的醇14(X-射线晶体表明,双环[3.1.0]己烷骨架与cryptotrione(1)相对构型相同)。用Dess-Martin高碘试剂氧化14能获得相应的酮,再使用LDA(1eq)在混合溶剂中(THF:DMPU=10:1)与溴化香叶酯进行单烷基化反应,从而获得单烷基化多烯酮15(非对映异构体混合物,柱色谱法不可分离),收率为72%(84% brsm)。由于化合物15不稳定且易于分解,作者试图通过将羰基还原为羟基来稳定酮15,由于羟基会降低α-质子的酸性,从而防止环化前体发生潜在的差向异构化。酮15在低温下被三仲丁基硼氢化锂(L-selectride)非对映选择性还原,以33%的收率(48%brsm)获得syn-C7,15′-β,β-多烯醇17作为主要非对映异构体(dr = 13:1)以及syn-C7,15′-α,α-多烯醇16为单一异构体,收率为35%。醇16进行氧化、差向异构化后,以88%的收率获得酮15(dr = 1:1)。为了稳定多烯醇17并防止其在酸性条件下发生阳离子-烯烃环化,作者以高收率将其羟基转化为乙酸酯(β-侧包含多烯和羟基)。
为了构建松香烷(Abietane)型骨架,作者探索了路易斯酸催化醇17的乙酸酯的多烯环化,形成反式十氢化萘产物18。根据Stork-Eschenmoser的规则,预计化合物17的C7-β-反式多烯乙酸酯的环化将以双椅式过渡态进行,获得具有C10-β-角甲基的反式萘烷产物18。当使用Bi(OTf)3催化剂时,可将17的乙酸盐转化为abietane型产品18。
随后作者开始将异丙基引入体系中,当使用NBS和AcOH时,可获得30%收率(50%brsm)的溴化物19。而在六氟异丙醇(HFIP)和DMF的混合溶剂中使用两当量的NBS时,收率可提高至53%(69%brsm)。随后,作者使用新开发的异丙基化条件对溴化物19进行异丙基化反应,但仅产生了还原性产物18和正丙基取代的产物。因此,作者尝试在Stoltz’s条件下逐步引入异丙基,如所预期一致,溴化物19与异丙烯基三氟硼酸钾可顺利地进行Suzuki-Miyaura偶联反应,从而生成烯烃20,收率为80%(约3:1)。使用多种经典方案对烯烃20进行直接氢化的尝试完全失败,可能是由于乙酰基和新戊酰基的空间位阻所致。当用DIBAL-H还原20以减轻该空间位阻,随后进行氢化反应,成功地以91%的收率得到了二醇21。

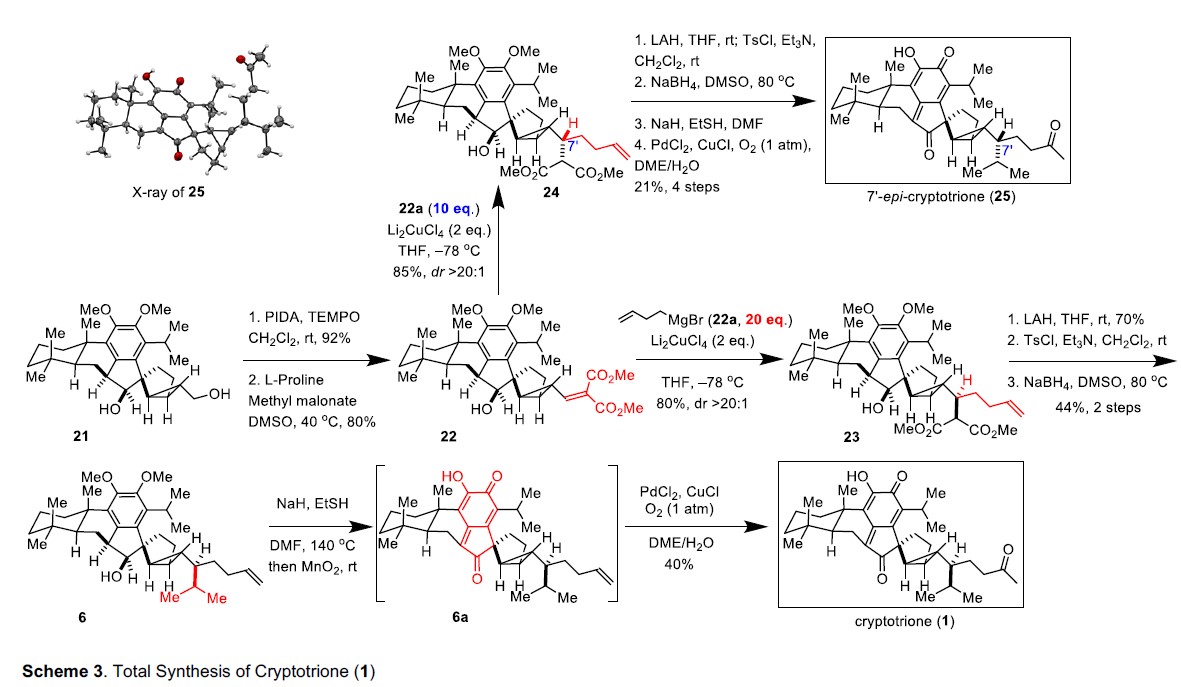
Cryptotrione(1)和 7’-epi-cryptotrione(25)的合成(Scheme 3)
在获得上述二醇21之后,作者开始对侧链的引入进行了研究。使用PIDA-TEMPO对21的伯羟基进行选择性氧化,随后与丙二酸二甲酯进行Knoevenagel缩合反应,获得80%收率的α,β-不饱和丙二酸22。当在-78℃下使用烯丙基溴化镁(22a,20eq)和Li2CuCl4(2eq)作为铜源时,所需的共轭加成产物23的收率为80%,dr>20:1。有趣的是,在-78℃下,使用不同的试剂,在烯丙基溴化镁(22a,10eq)和Li2CuCl4(2当量)时,共轭物加成的立体选择性完全相反,形成具有出色的dr(在C7’处> 20:1)的24(C7′-23的顶点)。因此,由于形成了不同的铜镁配合物,可以使用不同量的烯丙基溴化镁和加成模式来完全控制C7’处的立体化学。
因此,由迈克尔受体22衍生的烯烃23和24可分别合成cryptotrione(1)和 7’-epi-cryptotrione(25)。在23使用LiAlH4还原,选择性甲苯磺酸化和氢化物取代所得的甲苯磺酸盐之后,3步反应中以30%的收率生成烯烃6。然后在碱性条件下将6脱甲基,得到二羟基中间体,不幸的是,该中间体在空气中不能自动氧化成其相应的邻位醌。取而代之的是,该二羟基苯酚中间体用MnO2氧化,生成了对亚甲基醌化合物6a,将其直接进行Wacker氧化而无需进一步纯化,最终以40%的收率合成的cryptotrione(1)。此外,烯烃24经历从烯烃23到cryptotrione(1)的相似的步骤,以5步18%的收率实现了7′-epi-cryptotrione (25)的全合成。
总结
香港中文大学彭小水课题组首次以市售高藜芦酸(11)为初始底物,实现Cryptotrione (1)的全合成。同时该反应涉及三个关键步骤,即氯化铂(Ⅳ)催化烯炔环异构化构建双环[3.1.0]己烷骨架,路易斯酸引发多烯环化构建松香烷(Abietane)型三环二萜骨架,在侧链非对映共轭加成实现叔碳中心的控制。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.