

概要
1978年,日本Hokkaido大学药学部(Faculty of Pharmaceutical Sciences, Hokkaido University, 北海道大学薬学部)的Yonemitsu(米光 宰, Yonemitsu Osamu)研究室首次报道了使用有机催化剂脯氨酸实现Meldrum酸、乙醛与吲哚之间的三组分一锅缩合反应,成功完成3-取代吲哚衍生物的合成[1]。
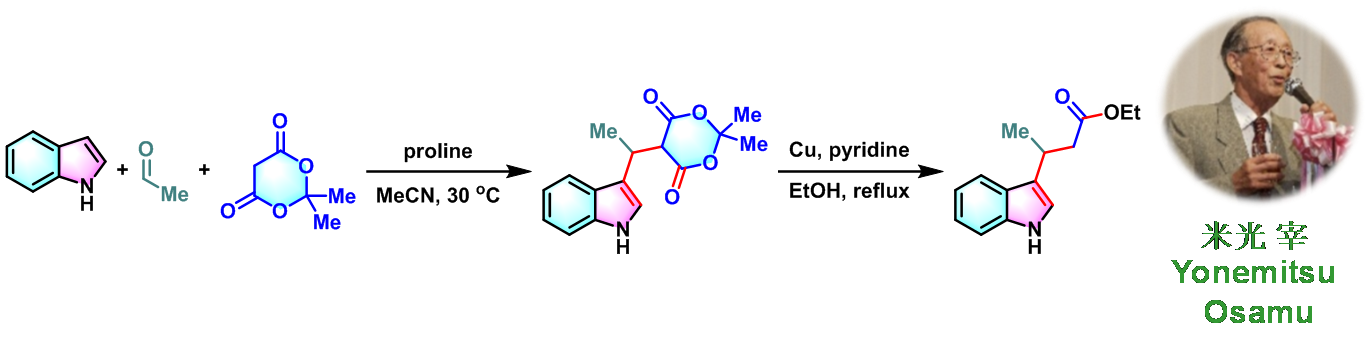
1982年,Yonemitsu对上述三组分缩合反应进行了进一步研究,并将底物范围扩展至各类脂肪醛与芳香醛,以较高产率获得一系列5-(1H-吲哚-3-基烷基)-2,2-二甲基-1,3-二噁烷-4,6-二酮类化合物,并在铜催化下进一步转化,从而获得各种取代的吲哚丙酸乙酯类化合物[2]。
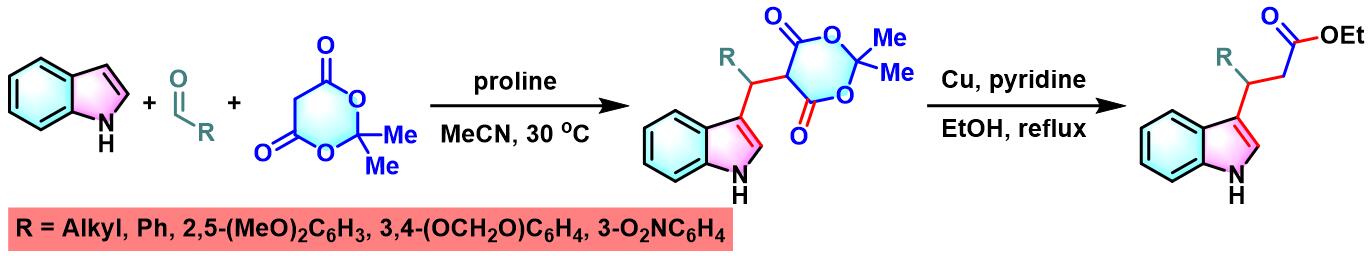
2005年,J.Sapi课题组采用手性的D-甘油醛衍生物及Garner 醛参与Meldrum酸、吲哚的立体选择性三组分一锅缩合反应,进而完成3,4-杂环环合的1-芳基取代四氢-β-咔啉(3,4-heterocycle-annulated 1-aryl-substitutetetrahydro-β-carboline)骨架的构建[3]。
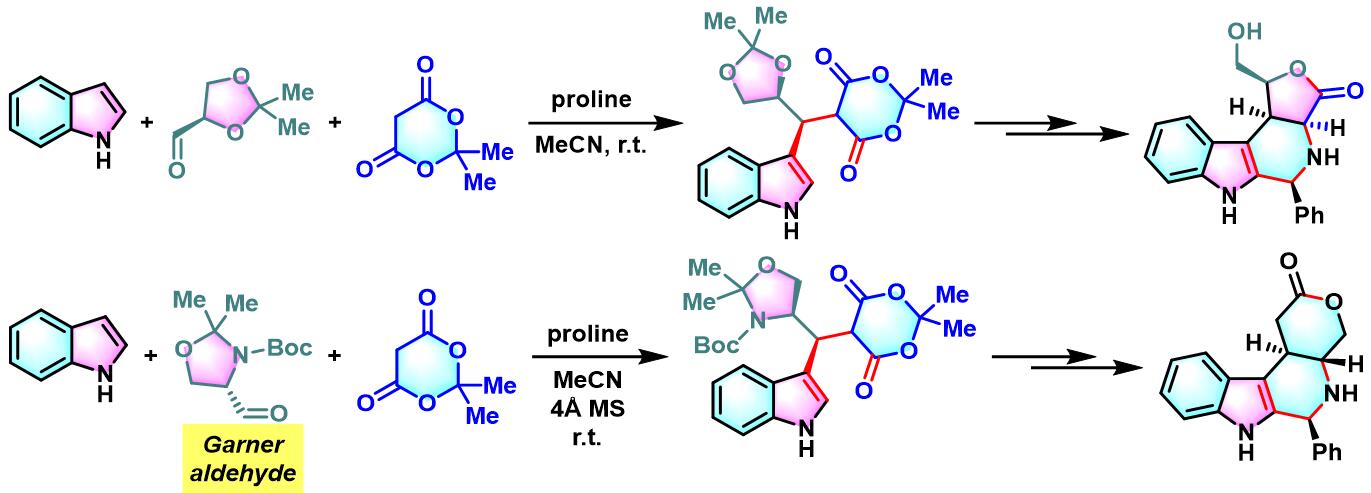
之后,该课题组对其它各类糖衍生的手性醛与吲哚、Meldrum酸之间三组分缩合进行了系统的研究,发现上述缩合过程具有良好的产率与较高的非对映选择性[4]。此外,作者还对反应机理进行了深入研究。

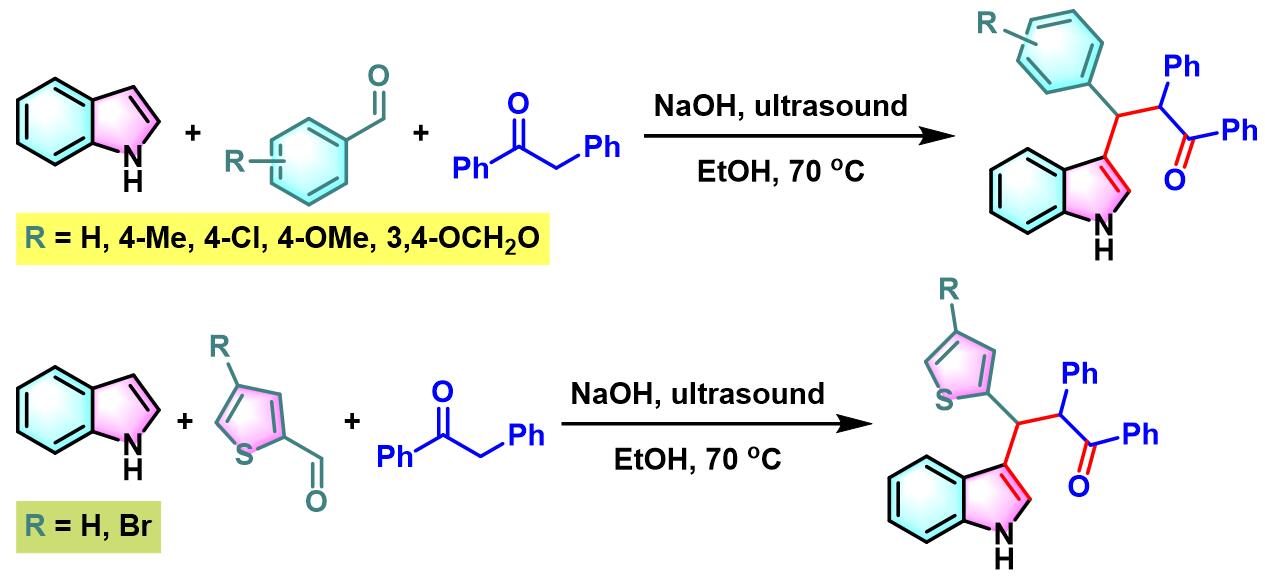
同时,S. Ji课题组通过超声辐射以及氢氧化钠作为催化剂的条件下,顺利实现吲哚与芳香醛或杂环芳香醛及去氧苯偶姻之间的三组分一锅缩合,从而完成β-吲哚基酮的合成[5]。
2007年,L.Liu与Y. Chen采用4 ÅMS及甲苯为溶剂的条件下,将底物范围进一步扩展至多聚甲醛与硝基乙酸酯[6]。
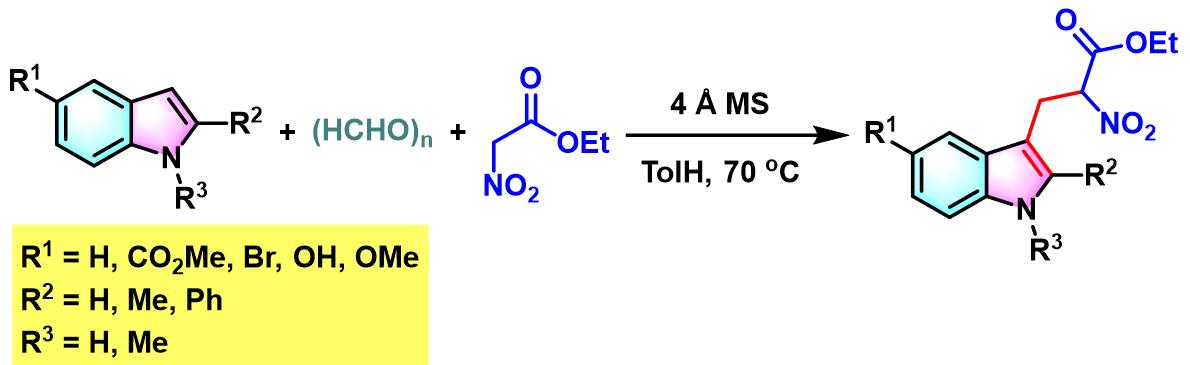
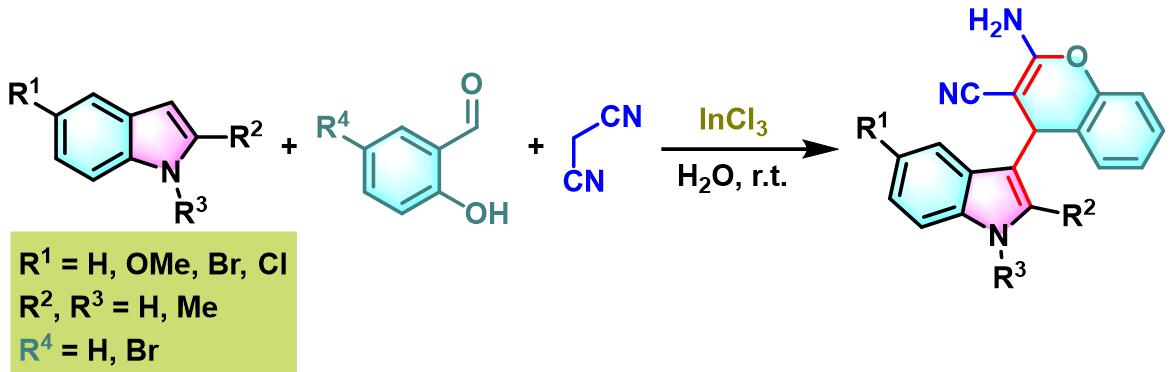
接下来,P. T. Perumal通过三氯化铟及水相条件下,将醛组分的应用范围扩展至各类羟基取代的芳香醛,并成功合成出一系列带有吲哚取代基的苯并吡喃类化合物[7]。
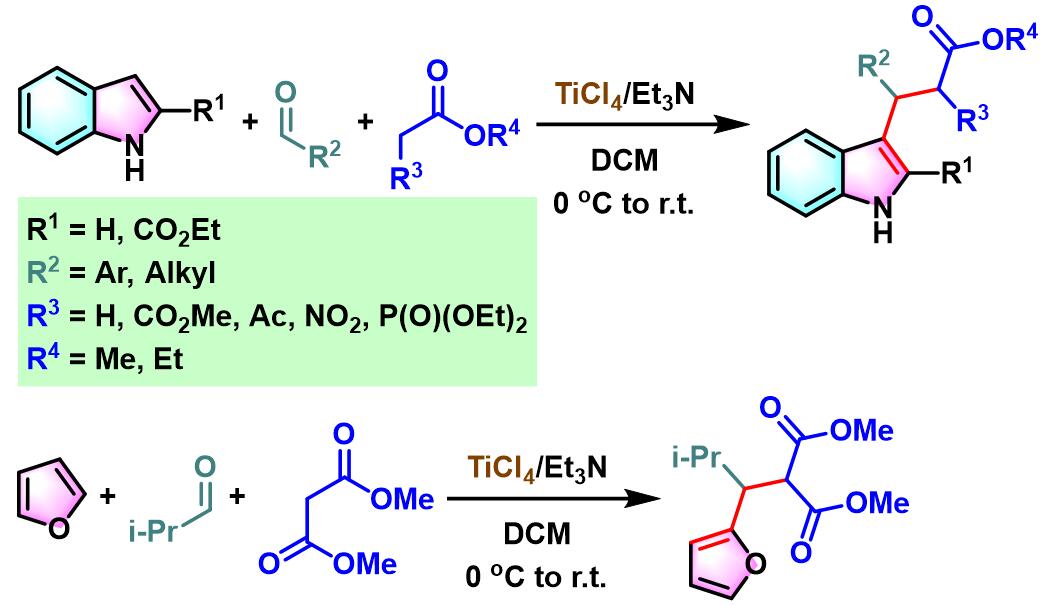
2008年,S. Gérard与J.Sapi研究发现,采用TiCl4/Et3N试剂,能够将底物范围扩展至其它取代吲哚或活泼亚甲基化合物[8],从而进一步拓宽了该方法学的应用范围。同时,S. Gérard与J.Sapi发现该试剂同样适用于呋喃、醛与丙二酸酯之间的三组分一锅反应[8]。
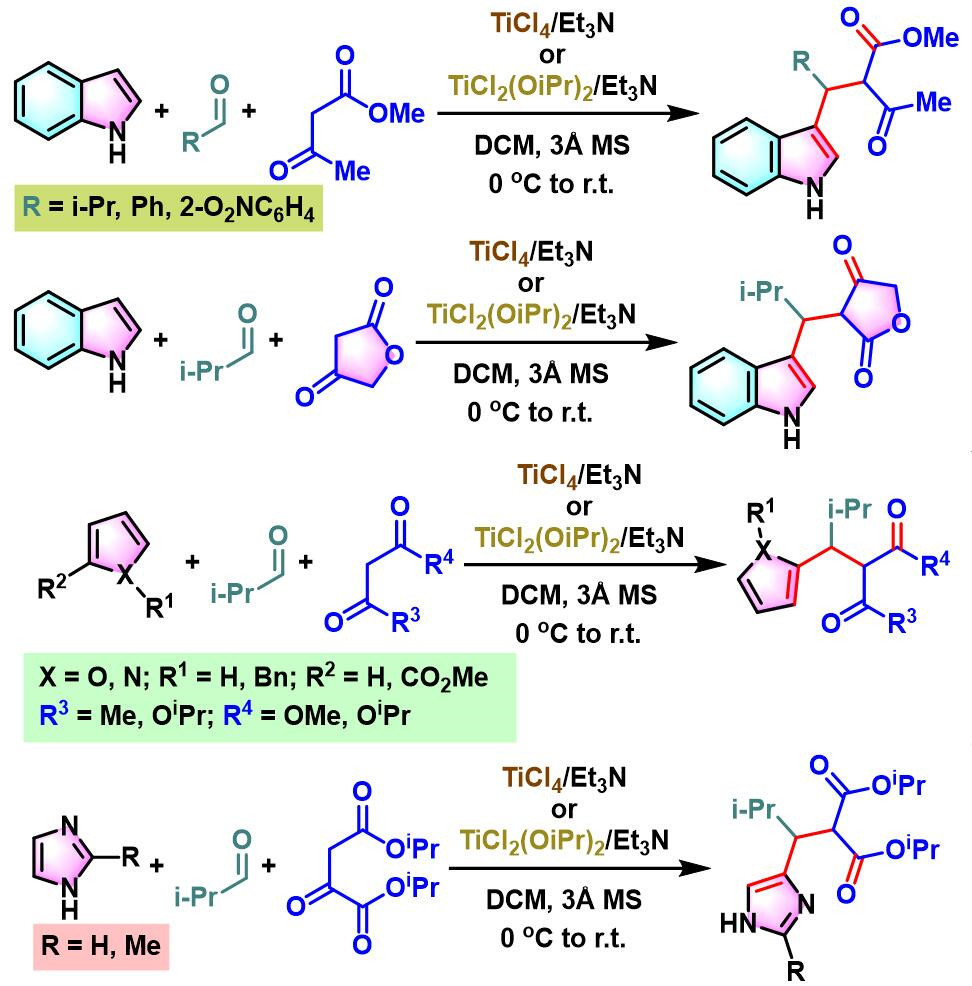
2010年,S. Gérard与J.Sapi研究表明,TiCl4 /Et3N或TiCl2(OiPr)2/Et3N试剂体系还能够进一步应用于呋喃、吡咯或咪唑与醛活泼亚甲基化合物之间的三组分缩合[9]。
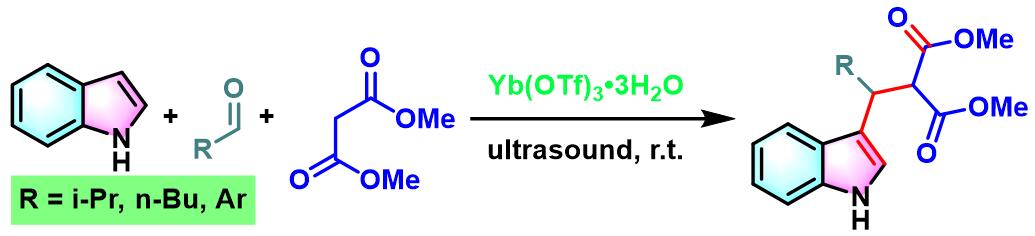
2011年,M.Curini通过Lewis酸(三氟甲磺酸镱)催化剂存,首次实现了醛、吲哚与Meldrum酸之间的无溶剂三分子缩合[10]。
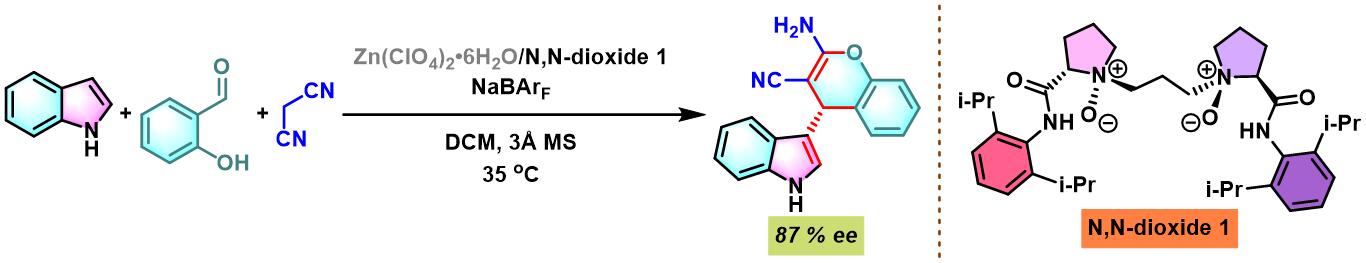
同时,X. Feng通过手性N,N-二氧化物与锌盐原位生成的N,N-二氧化物/锌(II)配合物催化剂,首次实现芳香醛、吲哚和丙二腈之间的对映选择性三组分一锅缩合[11]。

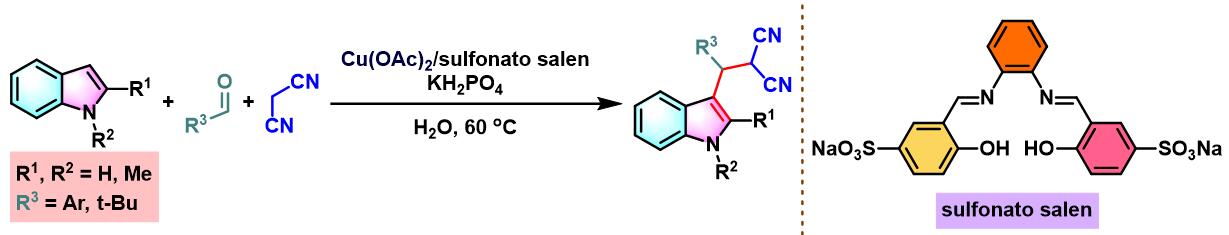
之后,X. Zhou发现采用原位形成的Cu-sulfonatosalen催化剂,同样能够顺利完成各类芳香醛与吲哚及丙二腈之间的三分子缩合[12]。
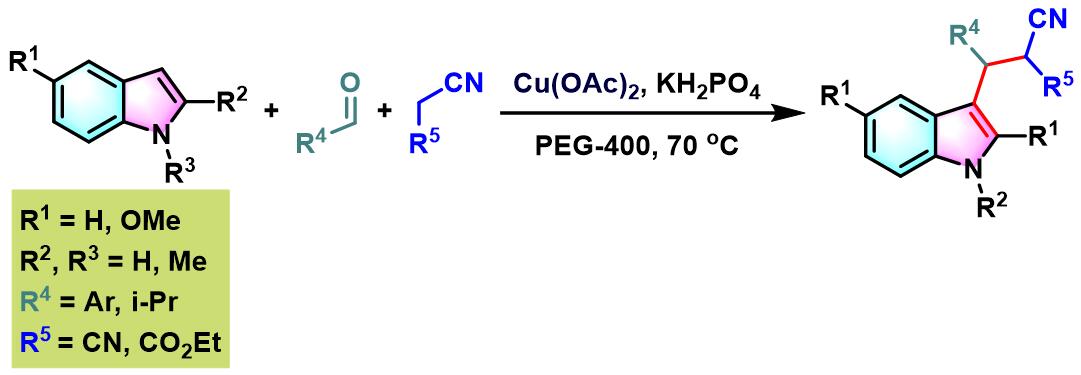
2012年,S.Chandrasekhar与R. Greé发现,在PEG-400及乙酸铜催化剂存在下,能够更为有效地实现各类芳香醛与吲哚及丙二腈或氰基乙酸酯之间的三组分缩合。反应过程无需额外添加其他配体[13]。
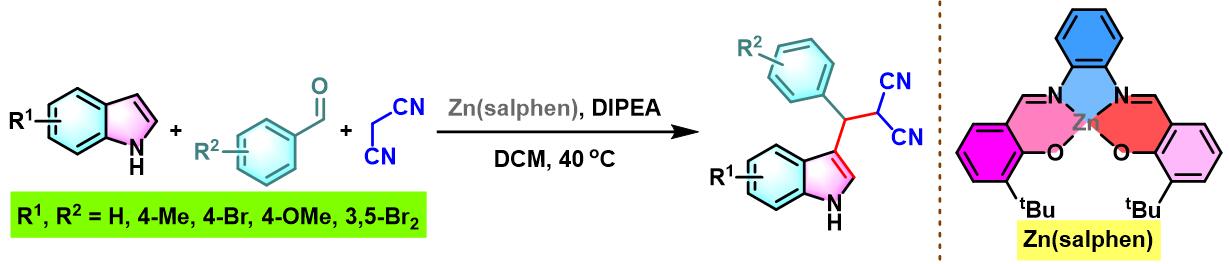
同时,A. W. Kleij研究表明,通过使用Zn(salphen)催化剂时,同样可以顺利完成芳香醛与吲哚及丙二腈之间的三组分缩合[14]。然而,仅获得中等程度的反应产率。
2013年,Y. Wan研究发现,采用PEG-200能够极大地促进芳香醛与吲哚及丙二腈之间的三组分一锅缩合。该条件更加温和且环境友好,同时,能够获得优良的产物收率[15]。
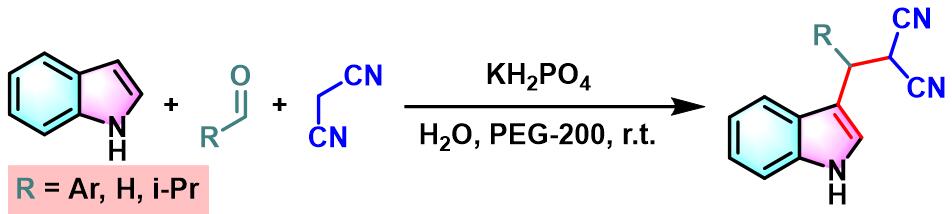
此外,K. N.Singh研究表明,通过TBAF催化剂,同样能够有效地完成芳香醛与吲哚及各类活泼亚甲基化合物在无溶剂条件下的三组分一锅缩合[16]。
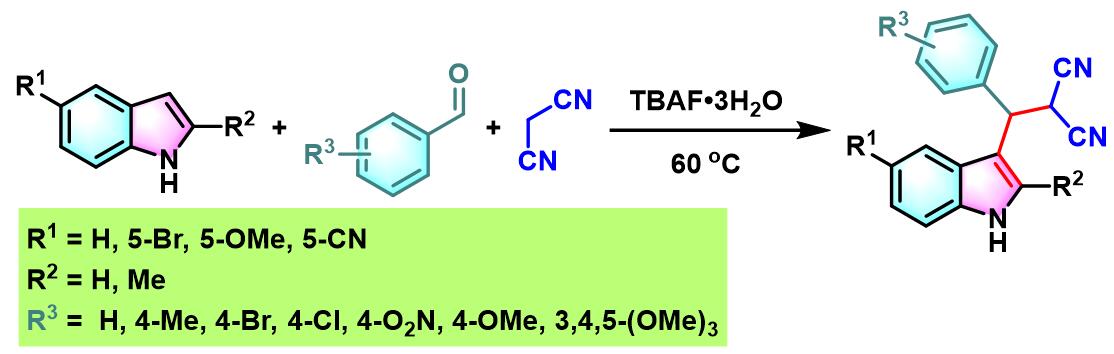
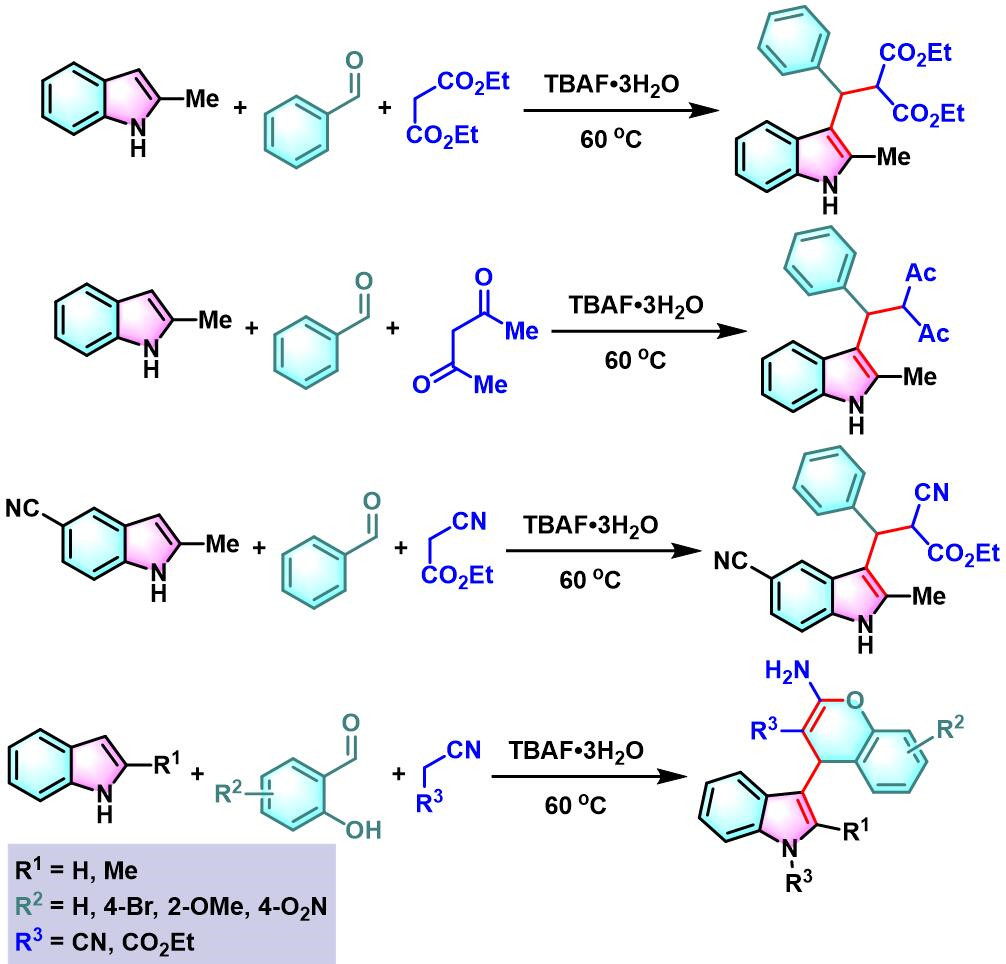
文献中将这种在碱或Lewis酸等催化剂存在下,亲核性杂环化合物 (如吲哚、呋喃、吡咯等) 与各类芳香醛、脂肪醛及活泼亚甲基化合物之间进行的三组分一锅缩合反应称为Yonemitsu三组分缩合(Yonemitsuthree component condensation)、Yonemitsu多组分反应(Yonemitsu multicomponent reactions)、Yonemitsu三分子缩合 (Yonemitsutrimolecular condensation)或Yonemitsu偶联 (Yonemitsu coupling)[1]-[2], [17]-[18]。该反应具有温和的反应条件,良好的区域与立体选择性、优良的产率、广泛的底物范围等优点。同时,反应后处理过程较为简便。目前,该反应已经广泛应用于各类亲核性杂环化合物的区域选择性官能团化[19]-[22]及各类生物活性分子[23]的合成设计。因篇幅限制,对于该反应的近期研究进展,小编将在下一期进行介绍。
基本文献
- [1] Y. Oikawa, H. Hirasawa, O. Yonemitsu, Tetrahedron Lett. 1978, 20, 1759. doi: 10.1016/0040-4039(78)80037-9.
- [2] Y. Oikawa, H. Hirasawa, O. Yonemitsu, Chem. Pharm. Bull. 1982, 30, 3092. doi: 10.1248/cpb.30.3092.
- [3] E. Dardennes, Á. Kovács-Kulyassa, M. Boisbrun, C. Petermann, J.-Y. Laronze, J. Sapi, Tetrahedron Asymm. 2005, 16, 1329. doi: 10.1016/j.tetasy.2005.02.008.
- [4] E. Dardennes, S. Gerard, C. Petermann, J. Sapi, Tetrahedron: Asymmetry 2010, 21, 208. doi: 10.1016/j.tetasy.2009.12.017.
- [5] Z. Shen, S. Ji, S. Wang, X. Zeng, Tetrahedron 2005, 61, 10552. doi: 10.1016/j.tet.2005.08.042.
- [6] Y. Sui, L. Liu, J. Zhao, D. Wang, Y. Chen, Tetrahedron Lett. 2007, 48, 3779. doi: 10.1016/j.tetlet.2007.04.002.
- [7] G. Shanthi, P. T. Perumal, Tetrahedron Lett. 2007, 48, 6785. doi: 10.1016/j.tetlet.2007.07.102.
- [8] A. Renzetti, E. Dardennes, A. Fontana, P. de Maria, J. Sapi, S. Gérard, J. Org. Chem. 2008, 73, 6824. doi: 10.1021/jo800529q.
- [9] S. Gérard, A. Renzetti, B. Lefevre, A. Fontana, P. de Maria, J. Sapi, Tetrahedron 2010, 66, 3065. doi: 10.1016/j.tet.2010.02.025.
- [10] F. Epifano, S. Genovese, O. Rosati, S. Tagliapietra, C. Pelucchini, M. Curini, Tetrahedron Lett. 2011, 568. doi: 10.1016/j.tetlet.2010.11.128.
- [11] W. Chen, Y. Cai, X. Fu, X. Liu, L. Lin, X. Feng, Org. Lett. 2011, 13, 4910. doi: 10.1021/ol2019949.
- [12] Y. Qu, F. Ke, L. Zhou, Z. Li, H. Xiang, D. Wu, X. Zhou, Chem. Commun. 2011, 47, 3912. doi: 10.1039/C0CC05695B.
- [13] S. Chandrasekhar, V. Patro, G. P. K. Reddy, R. Greé, Tetrahedron Lett. 2012, 53, 6223. doi: 10.1016/j.tetlet.2012.09.008.
- [14] D. Anselmo, E. C. Escudero-Adán, M. M. Belmonte, A. W. Kleij, Eur. J. Inorg. Chem. 2012, 4694. doi: 10.1002/ejic.201200150.
- [15] L. Wang, M. Huang, X. Zhu, Y. Wan, Appl. Cat. A: General 2013, 454, 160. doi: 10.1016/j.apcata.2012.12.008.
- [16] N. Singh, B. K. Allam, D. S. Raghuvanshi, K. N. Singh, Adv. Synth. Catal. 2013, 355, 1840. doi: 10.1002/adsc.201300162.
- [17] J. Gerencsér, G. Dormán, F. Darvas, J. QSAR Comb. Sci. 2006, 25, 439. doi: 10.1002/qsar.200540212.
- [18] Y. Gu, Green Chem. 2012, 14, 2091. doi: 10.1039/C2GC35635J.
- [19] C. Nemes, L. J eannin, J. Sapi, M. Laronze, H. Seghir, F. Augé, J.-Y.Laronze, Tetrahedron 2000, 56, 5479. doi: 10.1016/S0040-4020(00)00446-4.
- [20] L. Jeannin, T. E. Nagy, E. Vassileva, J. Sapi, J.-Y. Laronze, Tetrahedron Lett. 1995, 36, 2057. doi: 10.1016/0040-4039(95)00180-K.
- [21] M. Boisbrun, L. Jeannin, L. Toupet, J.-Y. Laronze, Eur. J. Org. Chem. 2000, 3051. doi: 10.1002/1099-0690(200009)2000:17<3051::aid-ejoc3051>3.0.co;2-3.
- [22] C. Nemes, J.-Y. Laronze, Synthesis 1999, 254. doi: 10.1055/s-1999-3395.
- [23] C. Nemes, L. Jeannin, J. Sapi, M. Laronze, H. Seghir, F. Augé, J.-Y. Laronze, ARKIVOC 2004, 208. doi: 10.3998/ark.5550190.0005.717.
反应机理
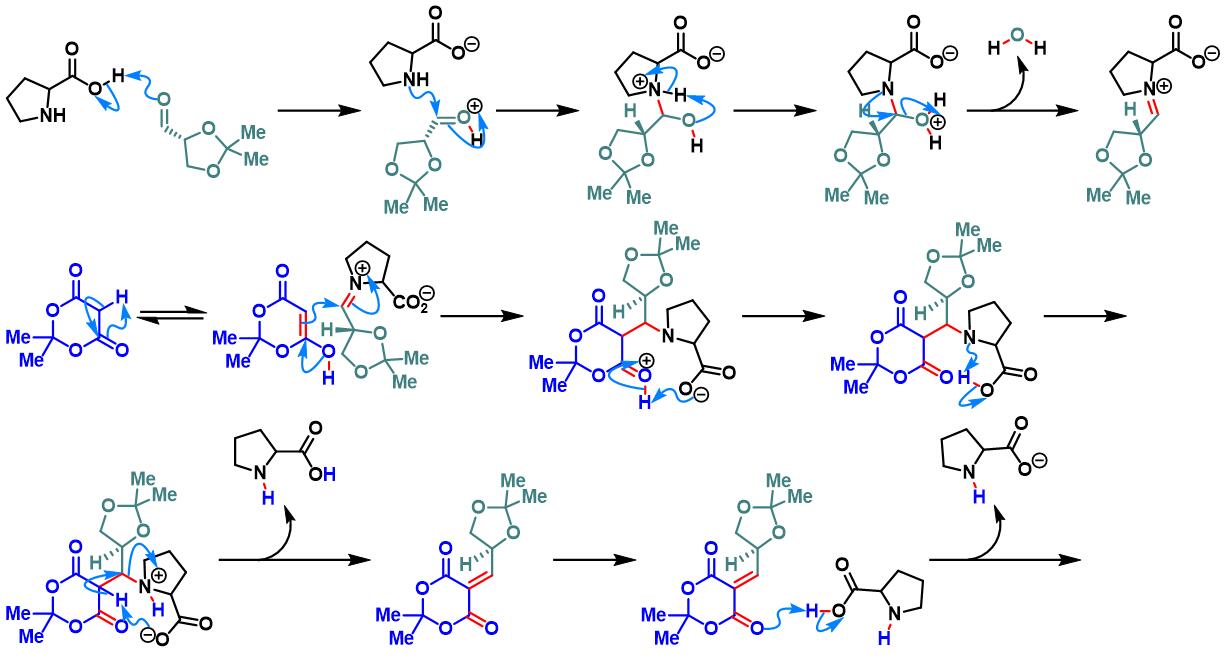
脯氨酸参与的Yonemitsu三组分缩合[1]
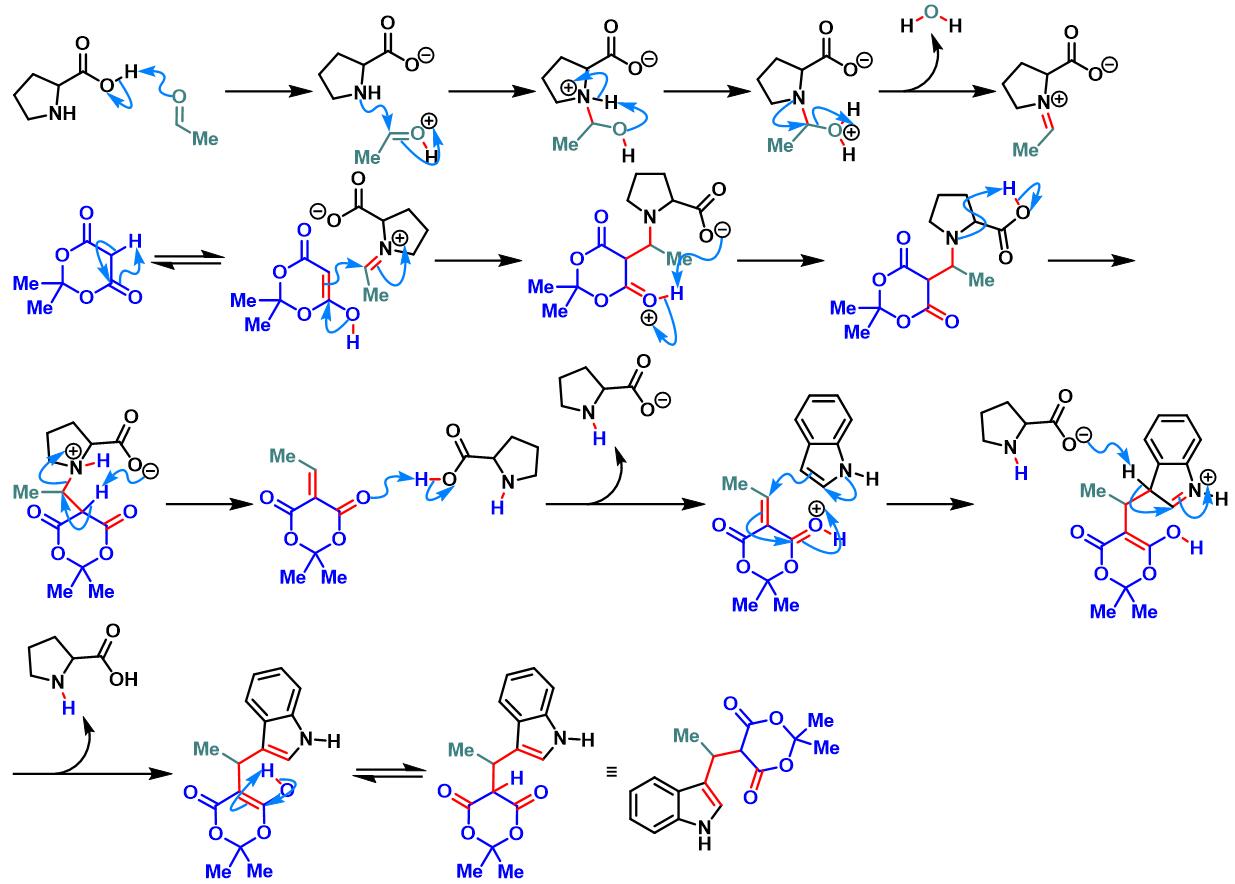
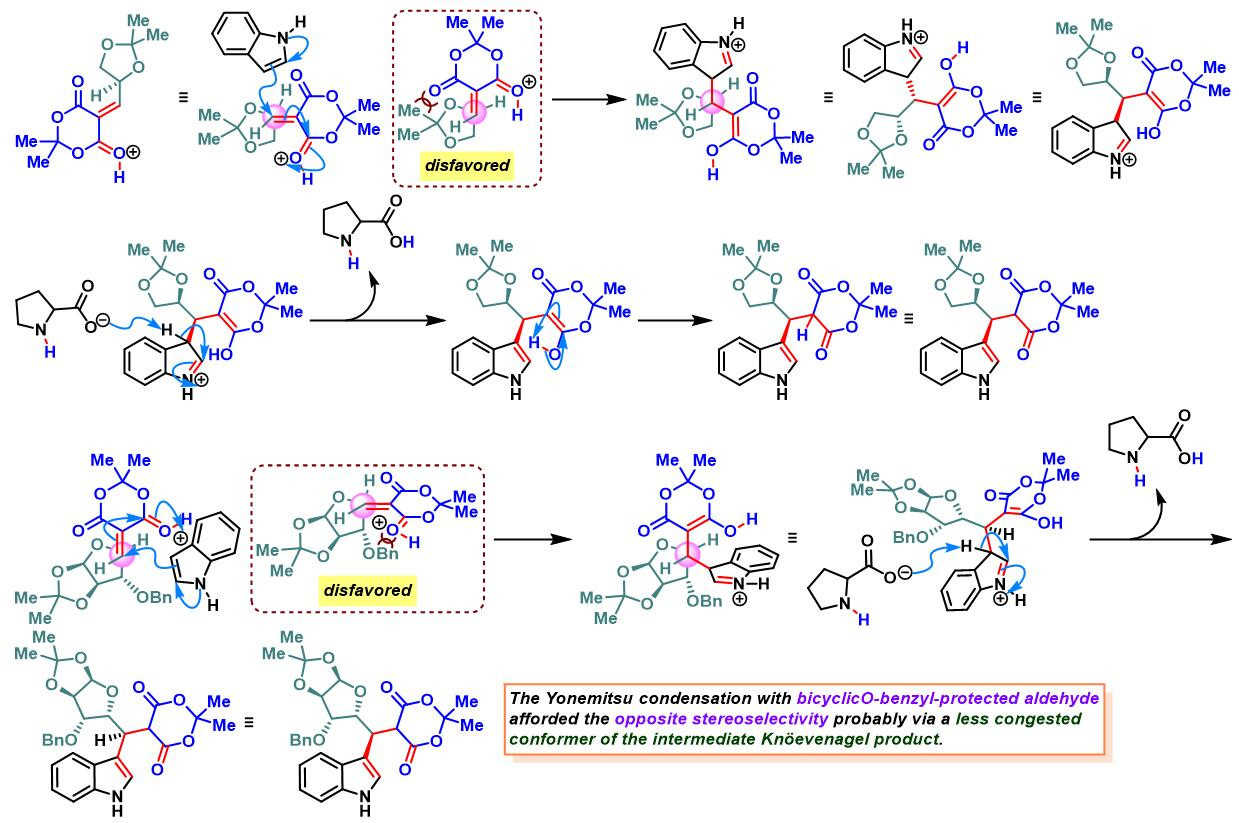
各类手性醛参与参与的Yonemitsu三组分缩合[2]-[5]
氢氧化钠催化的催化的Yonemitsu三组分缩合[6]

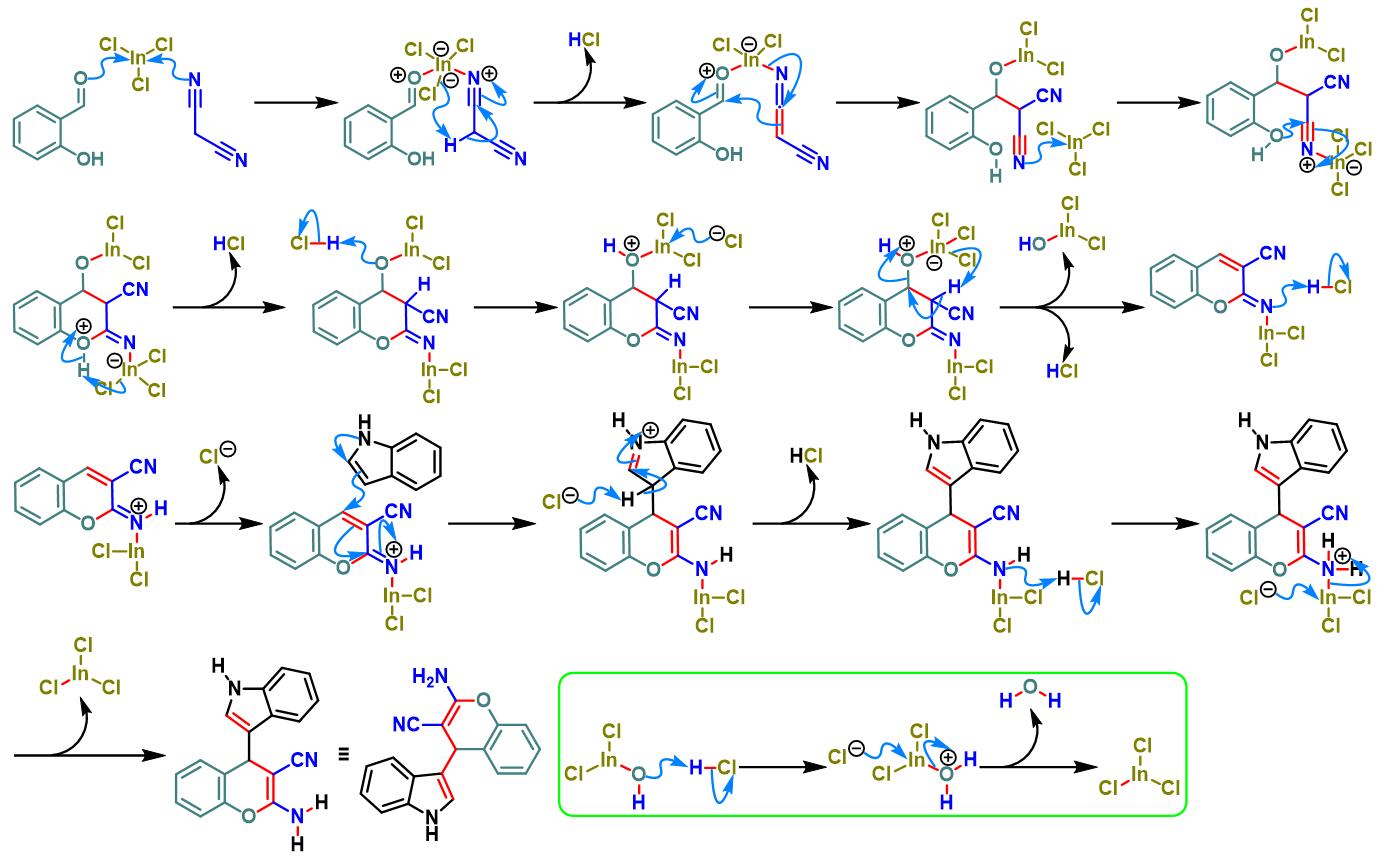
三氯化铟催化的Yonemitsu三组分缩合[7]
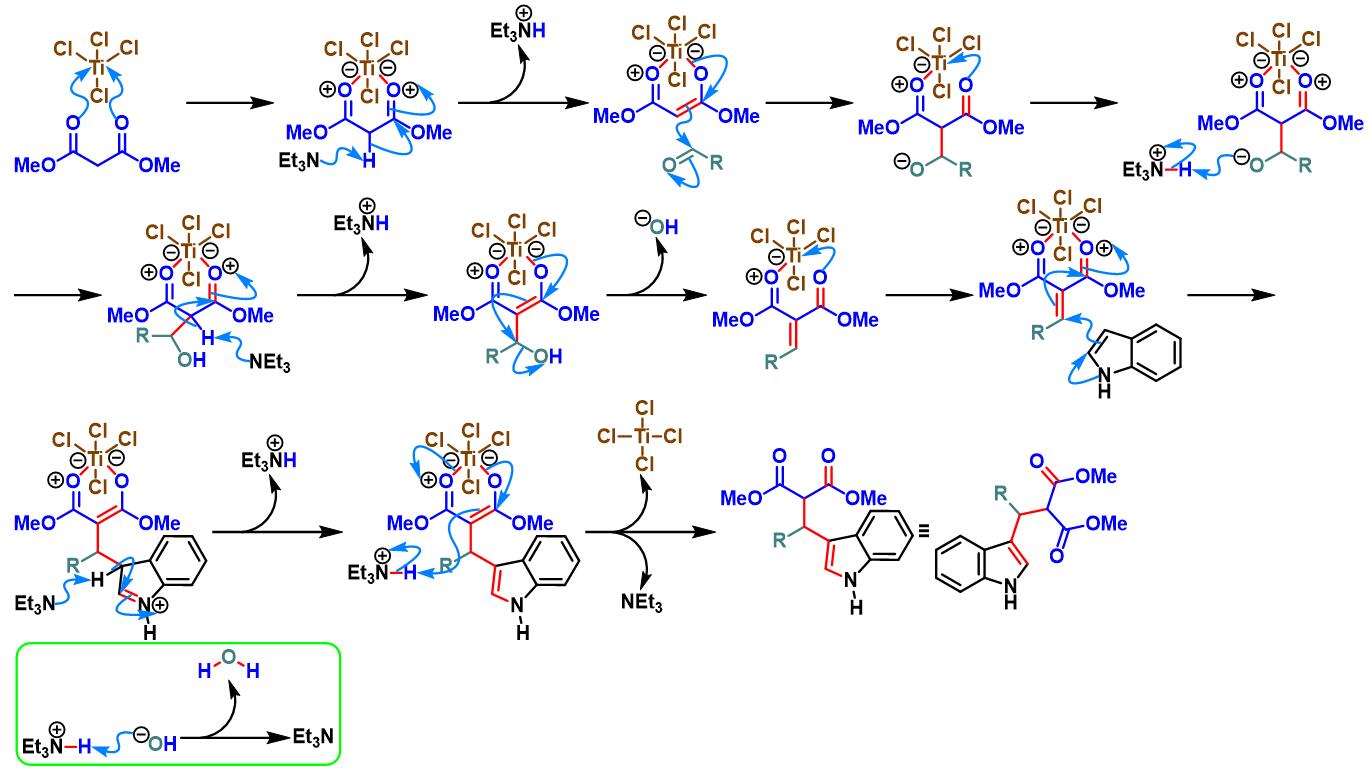
TiCl4/Et3N促进的Yonemitsu三组分缩合[8]-[11]
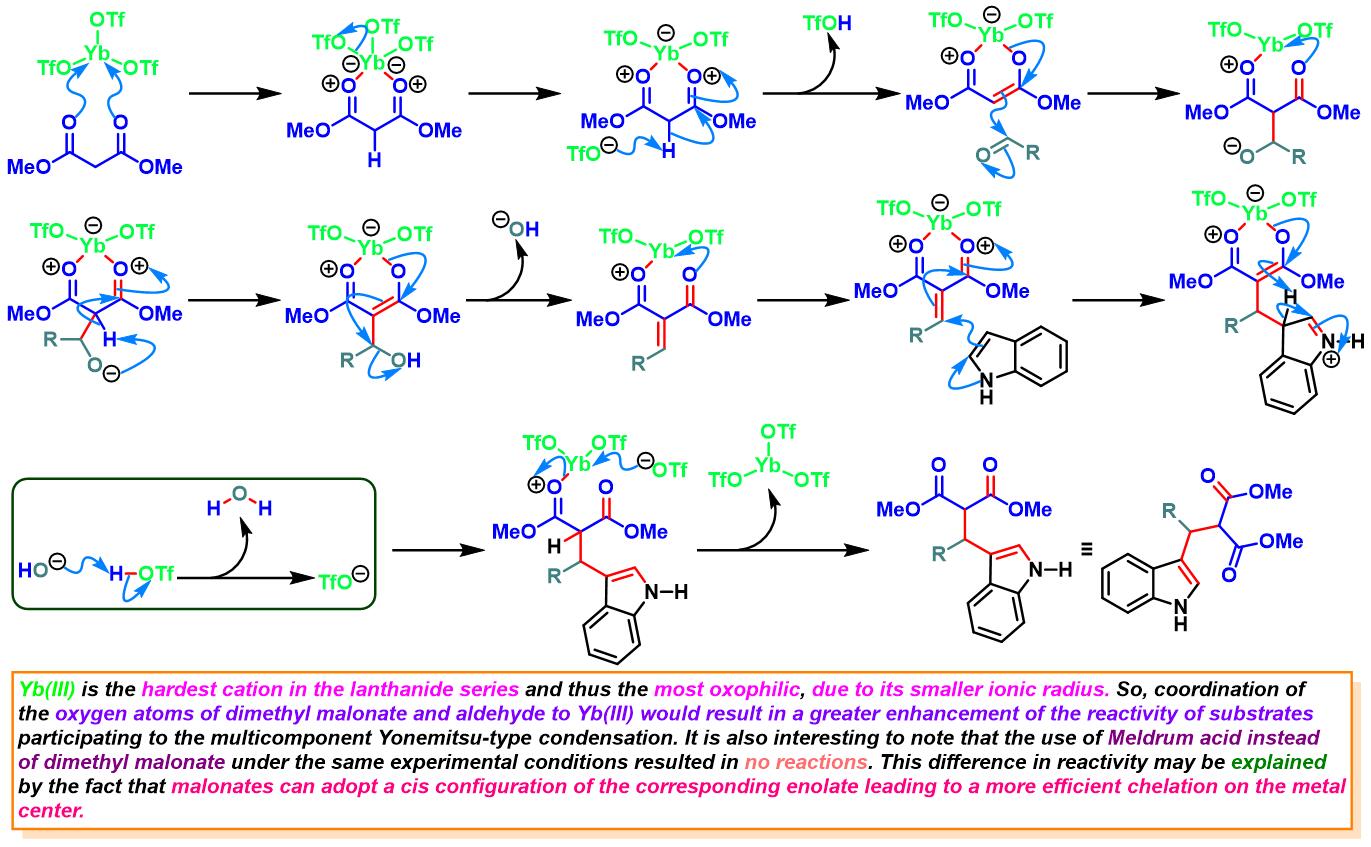
Yb(OTf)3催化的Yonemitsu三组分缩合[12]-[13]
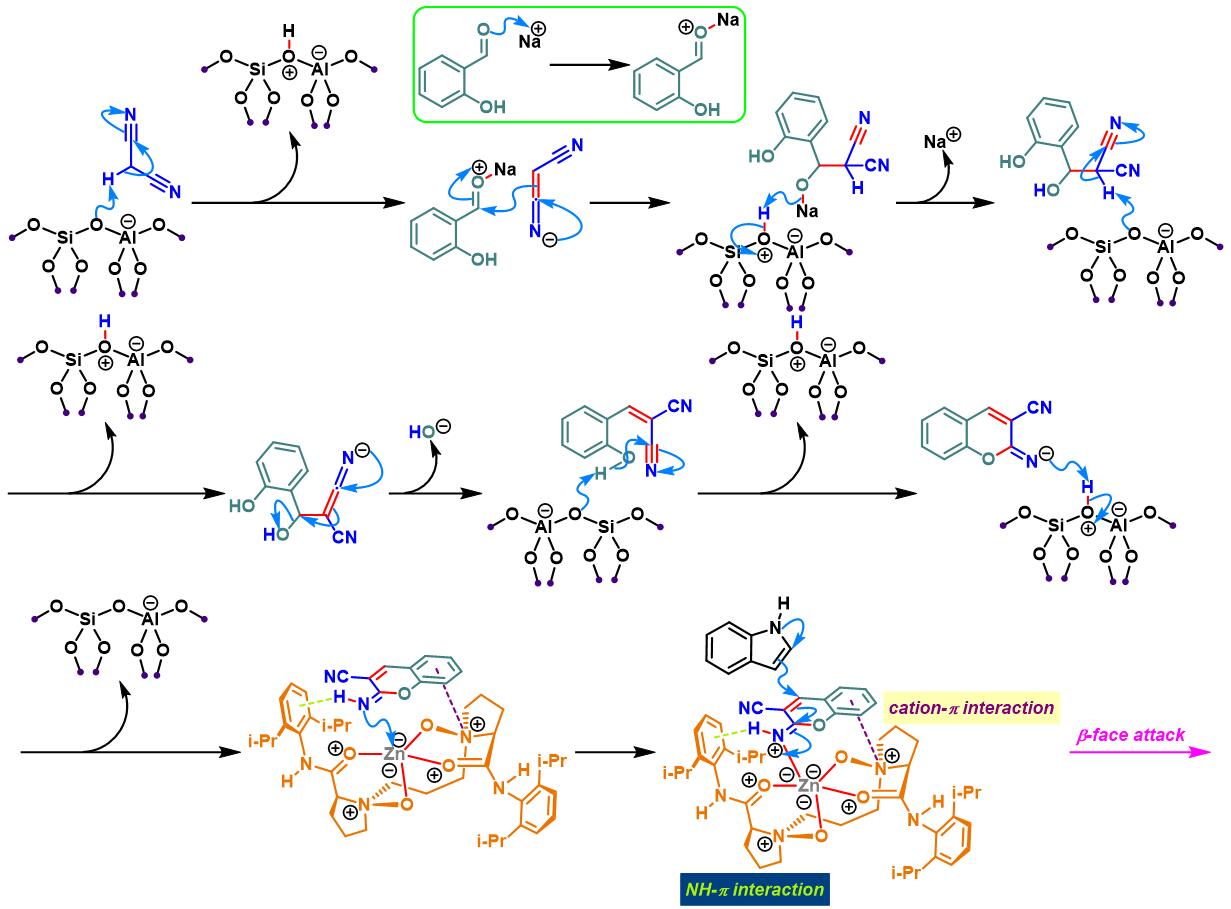
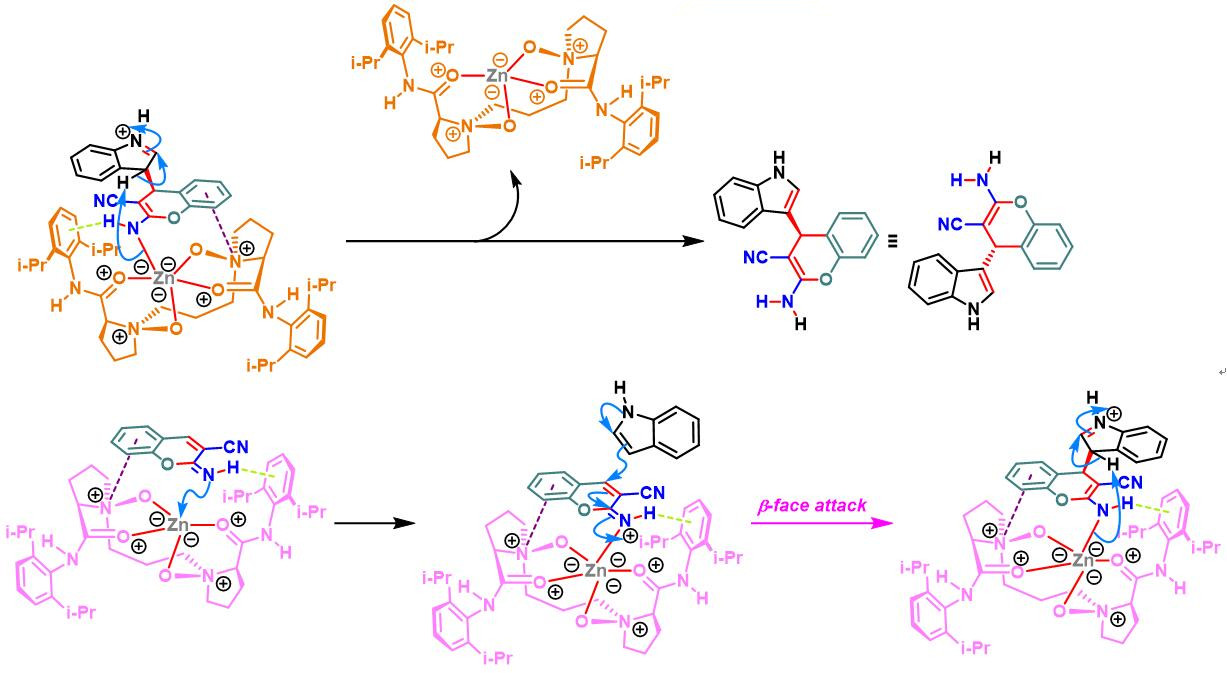
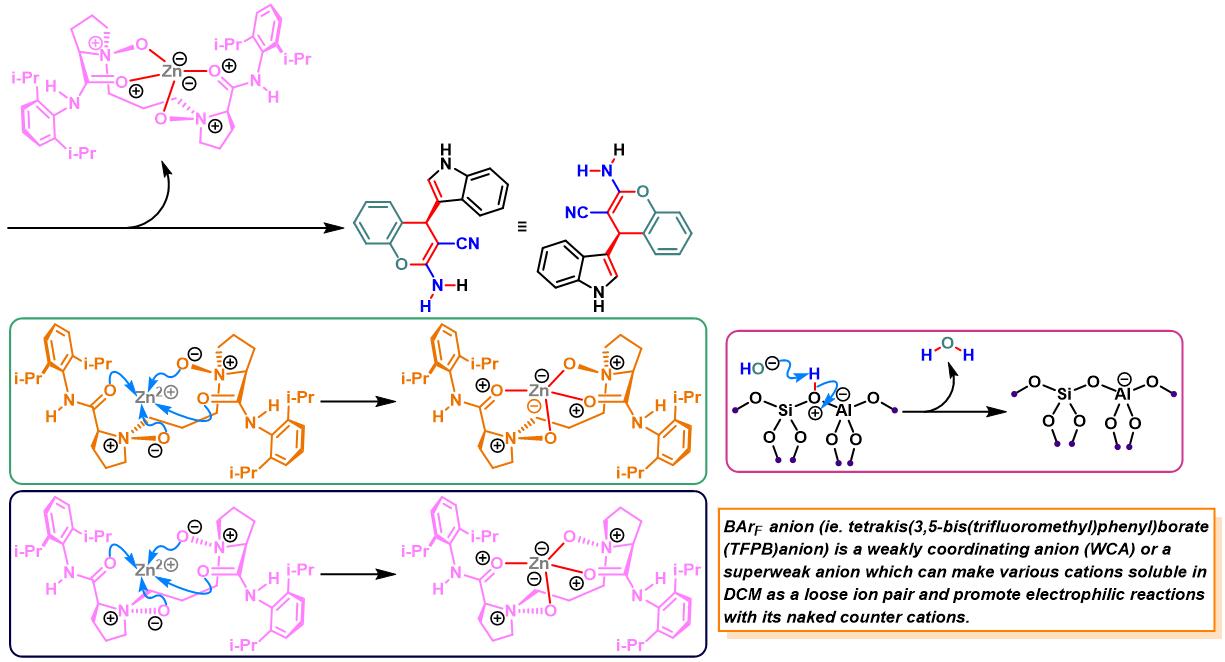
N,N–二氧化物-Zn(II)配合物催化的Yonemitsu三组分缩合[14]-[19]
乙酸铜-sulfonatosalen催化的Yonemitsu三组分缩合[20]-[22]

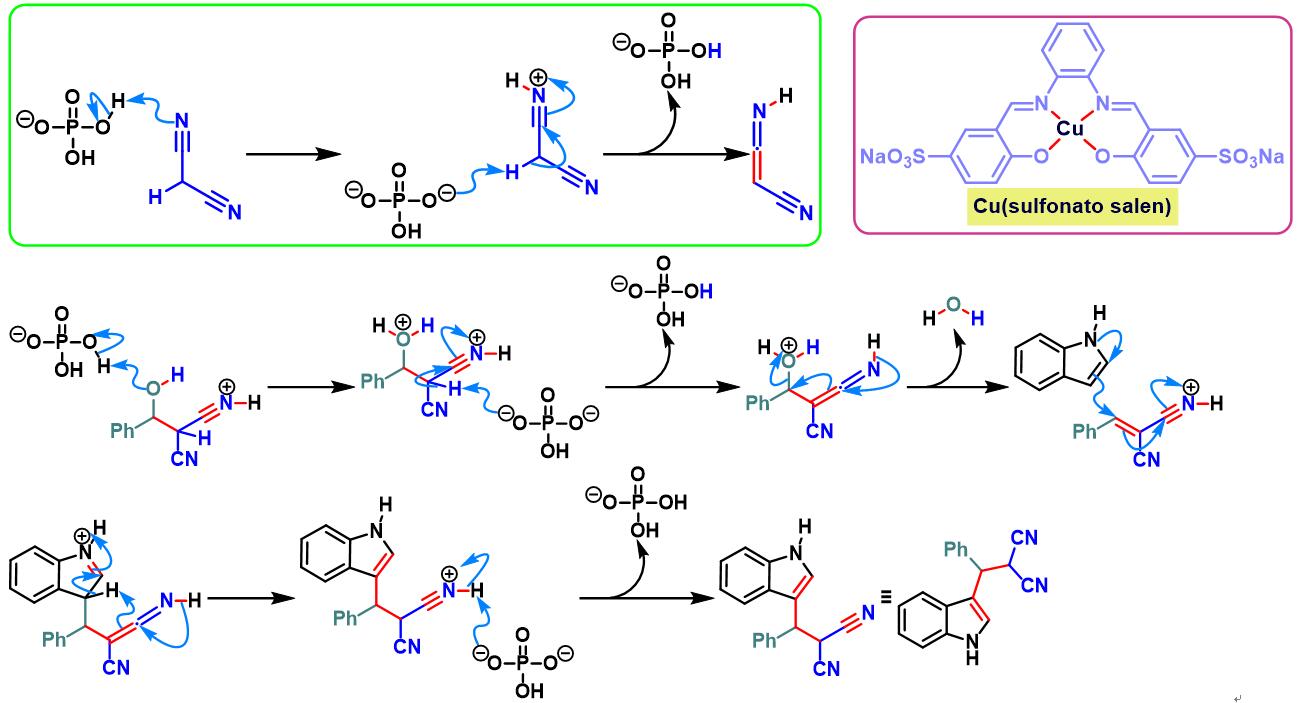
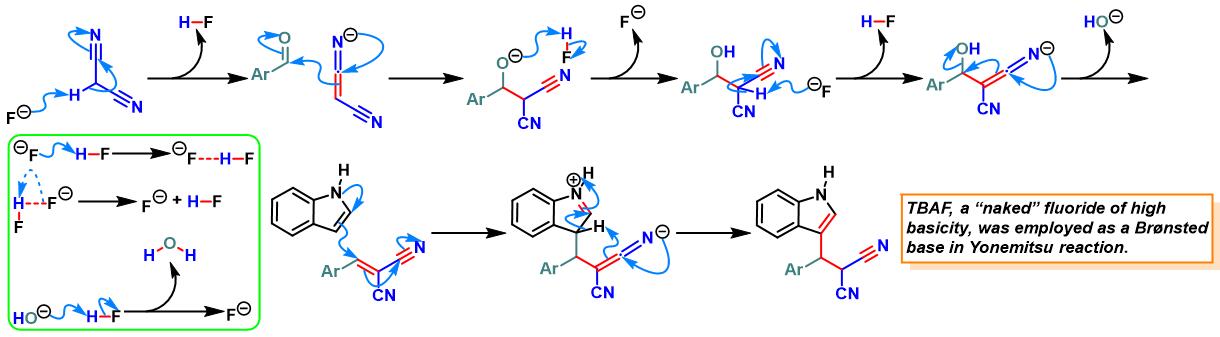
TBAF催化的Yonemitsu三组分缩合[23]-[26]
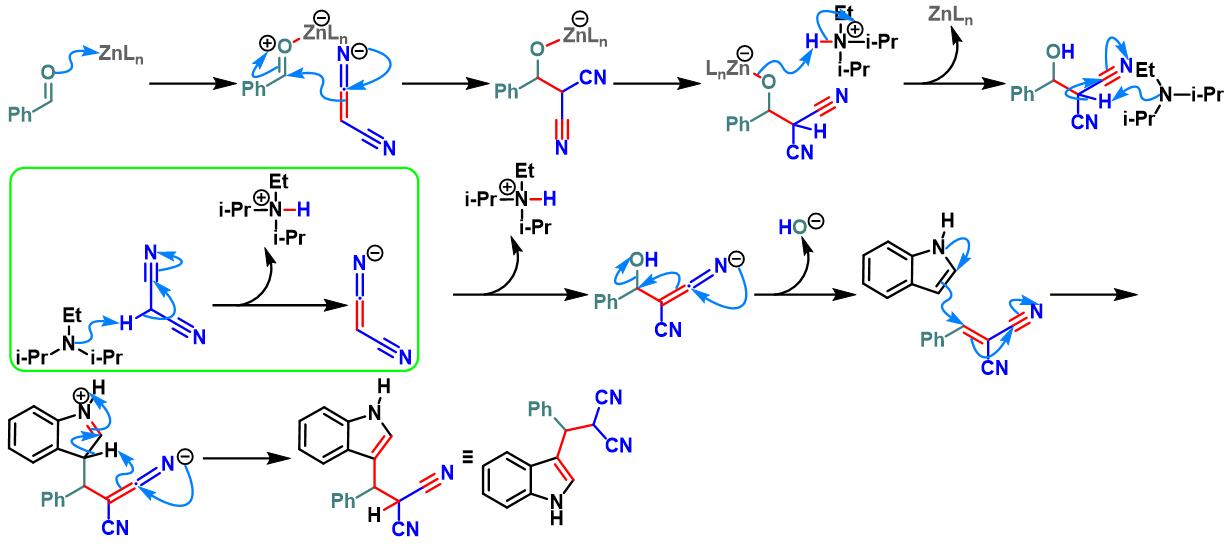
Zn(salphen)催化的Yonemitsu三组分缩合[27]
参考文献
- [1] M. Li, A. Taheri, M. Liu, S. Sun, Y. Gu, Adv. Synth. Catal. 2014, 356, 537. doi: 10.1002/adsc.201300790.
- [2] E. Dardennes, S. Gerard, C. Petermann, J. Sapi, Tetrahedron: Asymmetry 2010, 21, 208. doi: 10.1016/j.tetasy.2009.12.017.
- [3] M. Boisbrun, Á. Kovács-Kulyassa, L. Jeannin, J. Sapi, L. Toupet, J.-Y. Laronze, Tetrahedron Lett. 2000, 41, 9771. doi: 10.1016/S0040-4039(00)01719-6.
- [4] E. Dardennes, Á. Kovács-Kulyassa, A. Renzetti, J. Sapi, J.-Y. Laronze, Tetrahedron Lett. 2003, 44, 221. doi: 10.1016/S0040-4039(02)02537-6.
- [5] E. Muray, A. Alvarez-Larena, J. F. Piniella, V. Branchadell, R. M. Ortuño, J. Org. Chem. 2000, 65, 388. doi: 10.1021/jo991227j.
- [6] Z. Shen, S. Ji, S. Wang, X. Zeng, Tetrahedron 2005, 61, 10552. doi: 10.1016/j.tet.2005.08.042.
- [7] G. Shanthi, P. T. Perumal, Tetrahedron Lett. 2007, 48, 6785. doi: 10.1016/j.tetlet.2007.07.102.
- [8] A. Ma rrone, A. Renzetti, P. D. Maria, S. Gerard, J. Sapi, A. Fontana, N. Re, Chem. Eur. J. 2009, 15, 11537. doi: 10.1002/chem.200901595.
- [9] J. S. Yadav, S. Abraham, B. V. S. Reddy, G. Sabitha, Synthesis 2001, 2165. doi: 10.1055/s-2001-18068.
- [10] G. V. Karunakar, M. Periasamy, J. Org. Chem. 2006, 71, 7463. doi: 10.1021/jo060683m.
- [11] M. Periasamy, Arkivoc 2002, vii, 151. doi: 10.3998/ark.5550190.0003.717.
- [12] F. Bonadies, A. Lattanzi, L. R. Orelli, S. Pesci, A. Scettri, Tetrahedron Lett. 1993, 34, 7649. doi: 10.1016/S0040-4039(00)60424-0.
- [13] K. S. Pitzer, Acc. Chem. Res. 1974, 12, 271. doi: 10.1021/ar50140a001.
- [14] I. Krossing, I. Raabe, Angew. Chem. Int. Ed. 2004, 43, 2066. doi: 10.1002/anie.200300620.
- [15] X. Liu, L. Lin, X. Feng, Acc. Chem. Res. 2011, 44, 574. doi: 10.1021/ar200015s.
- [16] W. Wang, X. Liu, W. Cao, J. Wang, L. Lin, X. Feng, Chem.-Eur. J. 2010, 16, 1664. doi: 10.1002/chem.200902355.
- [17] K. Fujiki, S. Ikeda, H. Kobayashi, A. Mori, A. Nagira, J. Nie, T. Sonoda, Y. Yagupolskii, Chem. Lett. 2000, 62. doi: 10.1246/cl.2000.62.
- [18] S. H. Strauss, Chem. Rev. 1993, 93, 927. doi: 10.1021/cr00019a005.
- [19] B. Saha, S. De, S. Dutta, Catal. Sci. Technol. 2016, 6, 7364. doi: 10.1039/C6CY01370H.
- [20] J. Zhou, M. Ye, Z. Huang, Y. Tang, J. Org. Chem. 2004, 69, 1309. doi: 10.1021/jo035552p.
- [21] M. Bandini, M. Fagioli, P. Melchiorre, A. Melloni, A. Umani-Ronchi, Tetrahedron Lett. 2003, 44, 5843. doi: 10.1016/S0040-4039(03)01400-X.
- [22] J. Zhou, Y. Tang, J. Am. Chem. Soc. 2002, 124, 9030. doi: 10.1021/ja026936k.
- [23] N. Singh, B. K. Allam, D. S. Raghuvanshi K. N. Singh, Adv. Synth. Catal. 2013, 355, 1840. doi: 10.1002/adsc.201300162.
- [24] T. Ooi, K. Maruoka, Acc. Chem. Res. 2004, 37, 526. doi: 10.1021/ar030060k.
- [25] J. Hu, L. Liu, S. Yang, Y. Liang, Org. Biomol. Chem. 2011, 9, 3375. doi: 10.1039/C0OB01255F.
- [26] J. H. Clark, Chem. Rev. 1980, 80, 429. doi: 10.1021/cr60327a004.
- [27] U. C. Rajesh, V. S. Pavan, D. S. Rawat, ACS Sustain. Chem. Engin. 2015, 3, 1536. doi: 10.1021/acssuschemeng.5b00236.
反应实例
手性内酯的合成[1]

吲哚基苯并吡喃类化合物的合成[2]
吲哚螺环衍生物的合成[3]
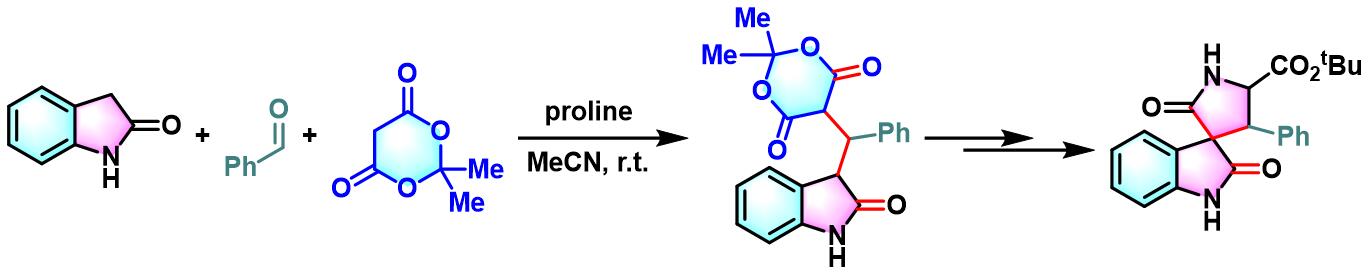
实验步骤
1. 脯氨酸参与的Yonemitsu三组分缩合
向吲哚(1 eq.)的乙腈溶液 (底物浓度为0.5 M)中加入醛(2 eq.)、脯氨酸(0.025-0.05eq.)与活泼亚甲基化合物(1 eq.)。将上述反应混合物在适宜温度下进行搅拌,直至反应结束。反应结束后,减压除去溶剂。将产生的晶体通过正己烷进行洗涤,即可获得相应缩合产物。
2. 手性醛参与参与的Yonemitsu三组分缩合
向手性醛(1 eq.)的乙腈溶液 (底物浓度为0.5 M)中加入吲哚(1.1 eq.)、脯氨酸 (0.01eq.)与Meldrum酸(1.1 eq.)。将上述反应混合物在室温与氮气气氛下进行搅拌,直至反应结束。反应结束后,减压除去溶剂。将粗产物通过硅胶柱色谱(环己烷/乙酸乙酯体系作为洗脱剂)分离纯化,获得相应缩合产物。
3. 氢氧化钠催化的催化的Yonemitsu三组分缩合
向反应烧瓶中加入吲哚(1eq.)、去氧苯偶姻(1eq.)、醛(1eq.)与氢氧化钠(2 eq.)及乙醇(维持底物浓度为 0.2 M)。将上述反应混合物在70oC,超声波辐射条件下进行搅拌,并通过TLC监控,直至反应结束。反应结束后,加入适量50%乙醇水溶液进行搅拌。几分钟后,观察到有固体析出。随后,通过减压过滤,收集析出的固体。将上述固体通过重结晶 (95%乙醇作为溶剂)进行进一步纯化,获得相应缩合产物。
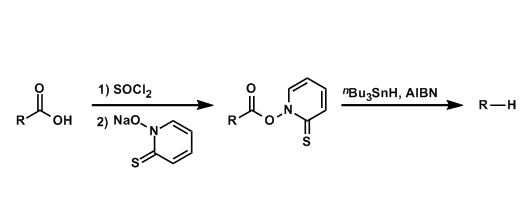
4. 三氯化铟催化的Yonemitsu三组分缩合
室温下,向反应烧瓶中加入水杨醛(1eq.)、丙二腈(1eq.)吲哚(1eq.)与水(维持吲哚浓度为 0.1 M)。再加入三氯化铟(0.02eq.)。将上述反应混合物在室温下进行搅拌,并通过TLC监控,直至反应结束。反应结束后,向上述反应混合物中加入乙酸乙酯进行萃取,再将合并的有机相通过无水硫酸钠干燥后,减压除去溶剂。粗产物通过硅胶柱色谱(正己烷/乙酸乙酯7:3v/v作为洗脱剂)分离纯化,获得相应缩合产物。
5. TiCl4/Et3N促进的Yonemitsu三组分缩合
0 oC,氮气气氛及3 Å分子筛存在下,将丙二酸酯(1eq.)加入至四氯化钛的无水DCM溶液中 (浓度为2 M),将产生的黄色悬浊液搅拌15 min。15 min后,加入三乙胺(1eq.),再将产生深红色均相溶液搅拌5 min。5 min后,再滴加醛(1eq.),继续在0oC下进行搅拌,直至深红色消失。最后,加入吲哚(1eq.),将上述反应混合物升至室温,继续搅拌,直至反应结束。反应结束后,加入1 M HCl淬灭反应。淬灭完成后,分离出有机相。将有机相通过无水硫酸镁进行干燥后,减压除去溶剂。将粗产物通过硅胶柱色谱或通过重结晶进行分离纯化,获得相应缩合产物。
6. 三氟甲磺酸镱催化的Yonemitsu三组分缩合
向反应烧瓶中加入吲哚(1eq.)、丙二酸二甲酯(1eq.)、醛(1eq.)与水合三氟甲磺酸镱(0.1eq.)。将产生的悬浊液在超声浴中进行震荡,并通过TLC(DCM/石油醚9:1v/v作为展开剂)监控,直至反应结束。反应结束后,加入1 M 氢氧化钠,滤除产生的沉淀。将合并的滤液通过乙醚进行萃取。合并的有机相通过无水硫酸镁进行干燥后,减压除去溶剂。粗产物通过硅胶柱色谱 (DCM作为洗脱剂)分离纯化,获得最终缩合产物。
7. 手性N,N–二氧化物/Zn(II)配合物催化的Yonemitsu三组分缩合
向反应烧瓶中加入手性N,N’-二氧化物 (0.2 eq.)、水合高氯酸锌 (0.1eq.)、NaBArF(0.2eq.)、丙二腈(1eq.)及DCM(维持丙二腈浓度为 0.02M)。将上述混合物在35 oC下搅拌30 min。之后,加入醛 (1eq.)。搅拌20 min后,再加入吲哚 (1.1 eq.)与适量的3 Å分子筛。将上述反应混合物继续在35 oC下进行搅拌,并通过TLC (DCM/石油醚 9:1v/v作为展开剂)监控,直至反应结束。反应结束后,将反应混合物通过硅藻土过滤,除去分子筛。再将合并的滤液进行减压浓缩后,获得粗产物。将粗产物通过硅胶柱色谱(石油醚/乙酸乙酯 2:1v/v作为洗脱剂)分离纯化,获得最终缩合产物。
8. 乙酸铜与sulfonatosalen催化的Yonemitsu三组分缩合
室温下向反应烧瓶中加入sulfonatosalen配体 (0.01 eq.)、乙酸铜 (0.01eq.)及水(维持乙酸铜浓度为 0.00033M)。将上述混合物搅拌2h。2h后,加入磷酸二氢钾 (1eq.)、丙二腈 (1.1eq.)、醛 (1eq.)及吲哚 (1.1 eq.)。将上述反应混合物在60oC下进行搅拌,并通过TLC监控,直至反应结束。反应结束后,将反应混合物冷却至室温,并加入乙酸乙酯进行萃取。将合并的有机相采用无水硫酸钠干燥后,减压除去溶剂。粗产物通过硅胶柱色谱 (石油醚/乙酸乙酯8:1v/v至4:1 v/v作为洗脱剂进行梯度洗脱)分离纯化,获得最终缩合产物。
9. TBAF催化的Yonemitsu三组分缩合
向反应烧瓶中加入吲哚 (1 eq.)、活泼亚甲基化合物 (1eq.)、醛 (1eq.)及TBAF•3H2O (0.1 eq.)。将上述混合物在60oC下进行搅拌,并通过TLC (DCM/石油醚 9:1v/v作为展开剂)监控,直至反应结束。反应结束后,将反应混合物冷却至室温,再加入适量水,同时,加入乙酸乙酯进行萃取。将合并的有机相采用无水硫酸钠干燥后,减压除去溶剂。粗产物通过硅胶柱色谱(正己烷/乙酸乙酯体系作为洗脱剂)分离纯化,获得最终缩合产物。
10. Zn(salphen)催化的Yonemitsu三组分缩合
向装有Zn-(salphen)催化剂 (0.05 eq.)、吲哚 (1.1 eq.)及丙二腈 (1.1 eq.)的反应烧瓶中加入DCM (维持吲哚浓度为0.18 M)。待上述混合物充分溶解后,再加入水杨醛 (1eq.) 与DIPEA (1eq.)。将上述反应混合物在40 oC下进行搅拌,并通过TLC监控,直至反应结束。反应结束后,将反应混合物冷却至室温,加入适量水,分离出有机相。将有机相通过无水硫酸钠干燥后,减压除去溶剂。粗产物通过硅胶柱色谱 (正己烷/乙酸乙酯 8:1 v/v至4:1 v/v作为洗脱剂进行梯度洗脱)分离纯化,获得最终缩合产物。
参考文献
- [1] E. Dardennes, S. Gerard, C. Petermann, J. Sapi, Tetrahedron: Asymmetry 2010, 21, 208. doi: 10.1016/j.tetasy.2009.12.017.
- [2] W. Chen, Y. Cai, X. Fu, X. Liu, L. Lin, X. Feng, Org. Lett. 2011, 13, 4910. doi: 10.1021/ol2019949.
- [3] F. Cochard, M. Laronze, E. Prost, J.-M. Nuzillard, F. Augé, C. Petermann, P. Sigaut, J. Sapi, J.-Y. Laronze, Eur. J. Org. Chem. 2002, 3481. doi: 10.1002/1099-0690(200210)2002:20<3481::aid-ejoc3481>3.0.co;2-c.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!









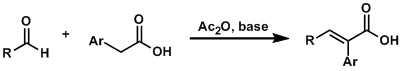







No comments yet.