
2017年,德克萨斯大学奥斯汀分校・Micharl J. Krische等人、成功开发出了通过Ru(0)催化剂催化的苯并丁烯酮与二醇的氢转移型环加成反应。由于该反应优异的位置・立体选择性,因此该反应被认为有望用作高效合成 type II polyketides。
“Ruthenium-catalyzed insertion of adjacent diol carbon atoms into C-C bonds:Entry to type II polyketides”
Bender, M.; Turnbull, B. W. H.; Ambler, B. R.; Krische, M. J.* Science 2017, 357, 779-781. DOI: 10.1126/science.aao0453
与以往的研究相比,优越性在于
对于扭曲的碳环为对象的C-C键活性化反应的研究其实从很早开始就已经在进行了。然而所用金属催化剂比如Pt 与Ni、Rh、Ru生成的metacycle集中在与π键的反应上。因此以饱和C-H键也就是σ(C-C)键为对象的插入反应的例子还很少。
在今天所介绍的论文中,通过使用氢转移型C-C偶联的概念来实现的对C-H的活化反应[1]。同时,得到的得到的稠环化合物具有桥头二醇结构,有望应用于高效合成type II polyketides。
技术手法的关键点
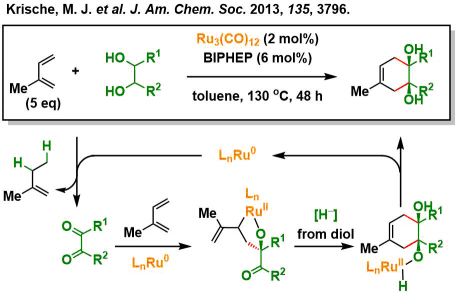
Krische课题组、通过使用烯烃和炔烃作为氢受体来加速Ru3(CO)12 催化剂对α-羟基酯和1,2-二醇的氧化、开发出了[4+2]型的环化反应等多种反应类型[2]。例如下图的反应[2b]所示、二醇被Ru(0)氧化形成二酮后、与二烯进行氧化偶联反应形成六元环。
主张的有效性验证
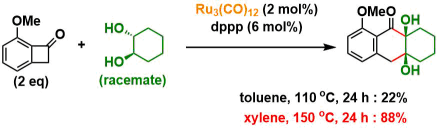
①反应条件的筛选优化
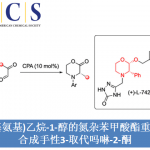
作者使用以下底物和甲苯溶剂进行初期探讨、在Ru3(CO)12 (2 mol%), dppp (6 mol%)存在下110 °C ,反应24 h后得到了目标环化产物,产率22%。二醇部位的立体选择性非常好,得到了syn only的立体选择性产物。然后通过调整反应溶剂,在使用二甲苯,150 °C 反应24h的条件下,产率提高到了88%。
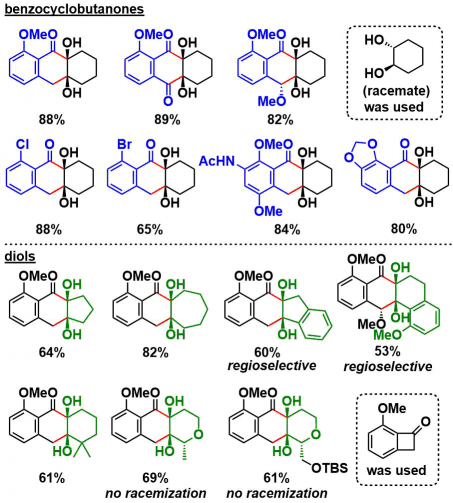
②底物拓展
对苯并环丁烯酮底物一侧进行screening、对于苯环上的取代基虽然仅仅局限于烷氧基与卤素,但是卤素有望可以进一步通过偶联用于合成多种衍生底物。
对于另一个底物,二醇来说、5 ~7元饱和碳环,邻位有二偕甲基的底物都可以顺利进行反应。此外,对于在α-位具有不对称碳的底物,反应进行而不会损害不对称性。
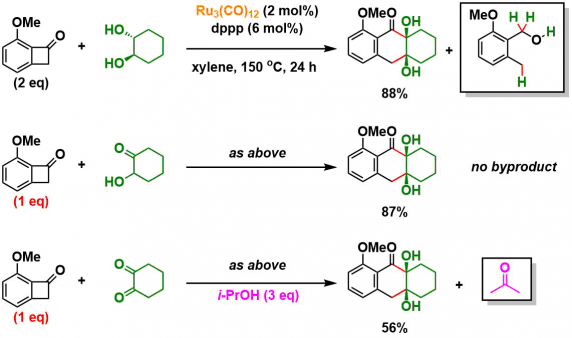
③反应机理的考察
在通常条件下,副产物中有苯并环丁烯酮原料开环后的副产物(2-甲氧基-6-甲基苯基)-甲醇的形成,这表明底物接受2当量的氢,氧化了底物二醇。另一方面,当使用酮醇作为原料时,由于体系转变为redox-neutral型,因此相对于苯并丁烯酮来说,酮醇的用量需要1当量。当二酮用作原料时,通过加入iPrOH作为还原剂进行反应[3]。
推定的反应机制如下。首先,原料彼此反应生成二酮底物。苯并环丁烯酮的C – C键被氧化加成到钌上形成钌烯酮。之后,通过连续的二酮加成反应得到二氧杂钌环。之后,通过原料供给氢,再从氢化钌中还原消除而生成目标物,再生Ru(0)。
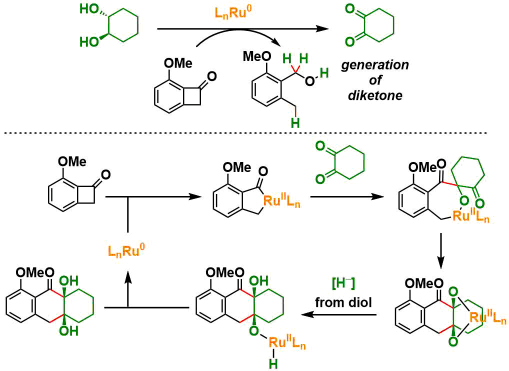
④手性反应的展开
作者利用上述standard的底物,通过使用(CF3CO2)2Ru(CO)(PPh3)2・MeOH与手性配体(R)-SEGPHOS进行反应、得到了51%ee的产物。到目前为止,这是唯一的一个手性合成的例子。
接下来需要关注的论文是?
- Ru 催化的C-C键活化,特别是用于手性催化反应领域的论文,还很少见。
参考文献
- Ketcham, J. M.; Shin, I.; Montgomery, T. P.; Krishe, M. J. Angew. Chem. Int. Ed.2014, 53, 9142. DOI: 10.1002/anie.201403873
- For example: (a) Leung, J. C.; Geary, L. M.; Chen, T.-Y.; Zbieg, J. R.; Krische, M. J. J. Am. Chem. Soc. 2012, 134, 15700. DOI: 10.1021/ja3075049 (b) Geary, L. M.; Glasspoole, B. W.; Kim, M. M.; Krische, M. J. J. Am. Chem. Soc. 2013, 135, 3796. DOI: 10.1021/ja400691t
- Johnson, T. C.; Totty, W. G.; Wills, M. Org. Lett. 2012, 14, 5230. DOI: 10.1021/ol302354z
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.