
本文作者:杉杉
导读
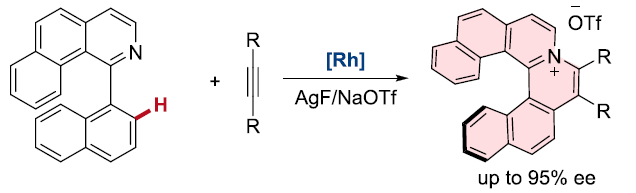
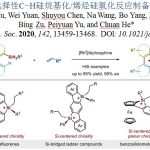
近日,上海有机所游书力研究员课题组在J. Am. Chem. Soc.发表论文,报道了通过铑(III)催化,实现1-芳基异喹啉衍生物与炔烃的对映选择性C-H活化/环化反应,从而以优良的收率(高达99%)与对映选择性(高达96%ee)获得一系列手性阳离子氮杂螺烯(azoniahelicene)衍生物。同时,作者对反应机理研究表明,C-H键的断裂步骤可能是转化限速步骤(turnover-limiting step)。
Enantioselective Synthesis of Azoniahelicenes by Rh-Catalyzed C-H Annulation with Alkynes
Q.Wang, W.Zhang, C. Zheng, Q. Gu, S. You
J.Am. Chem. Soc.ASAP. DOI: 10.1021/jacs.0c11735.
正文
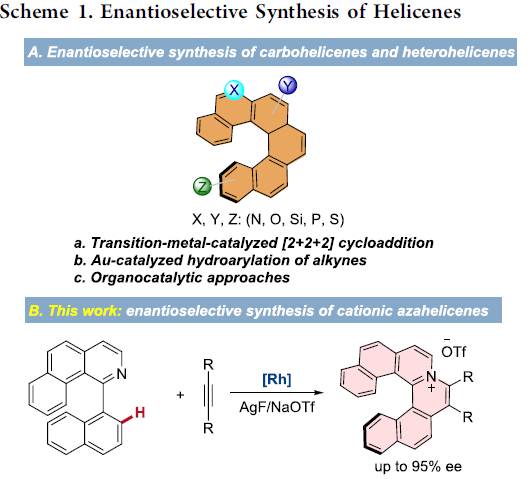
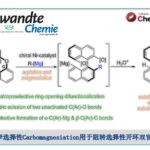
螺烯(helicenes),尤其是手性螺烯,由于具有邻位稠合的π共轭多环芳烃结构,已经广泛应用与诸多相关的研究领域,例如不对称催化、材料科学以及分子识别。在螺烯的催化对映选择性合成研究中(Scheme 1A),通过采用手性的Rh(I)、Ni(0)、 Co(I)以及Ir(I)配合物促进的炔烃的[2+2+2]环加成反应方法学,能够以中等至较高的对映选择性获得一系列手性螺烯分子。同时,Pd(0)配合物同样可以催化芳烃与炔烃的[2+2+2]环加成反应。而Au催化的炔烃对映选择性分子内氢芳基化反应方法学,同样能够应用于手性螺烯的构建。此外,有机催化的策略同样已经成功应用于手性螺烯分子的构建。然而,手性氮阳离子螺烯(azahelicenes)的不对称合成,却存在较大的挑战。目前已有的方法中,大多涉及外消旋体的拆分或手性HPLC分离。因此,迫切需要开发一种构建手性阳离子氮杂螺旋烯的催化不对称策略。目前,采用过渡金属催化的不对称C-H键官能团化,已经成为合成各类手性分子的有效方法,尤其采用Rh配合物参与的不对称C-H键官能团化更为有效。前期,本课题组已经报道通过采用手性环戊二烯基(Cp)配体配位的Rh(III)催化剂参与的1-芳基异喹啉衍生物的不对称C-H官能团化,从而完成一系列轴手性分子的合成,并获得中等至较高程度的对映选择性的 [1]。同时,受到Rh(III)催化的 1-芳基异喹啉衍生物与炔基化合物之间的C−H 活化/环化反应方法学相关研究的启发,作者设想,能否通过选择手性Rh(III)催化剂,实现1-芳基异喹啉衍生物与炔基化合物之间的对映选择性C-H活化/环化反应,从而获得对映体富集的手性阳离子氮杂螺旋烯(Scheme 1B)。
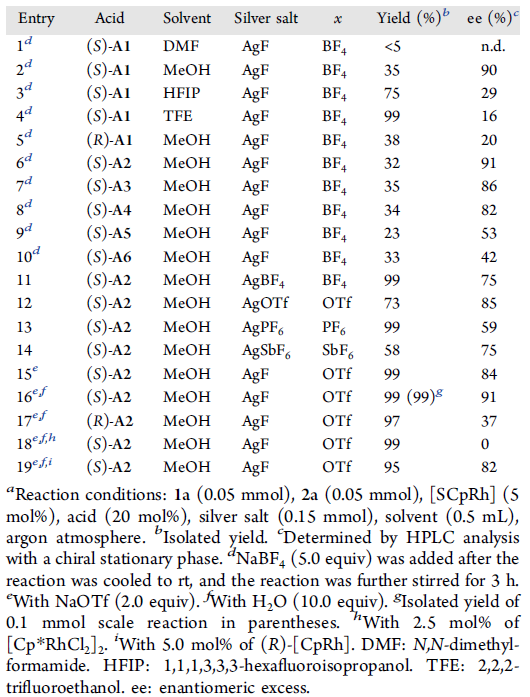
首先,作者选择1-芳基异喹啉衍生物1a与二苯乙炔2a作为模型底物,进行了相关反应条件的筛选(Table 1)。研究表明,反应的最佳条件为:采用(S)-[SCpRh] (5 mol%)作为催化剂,在(S)-A2(20 mol%)、AgF以及NaOTf存在下,甲醇溶剂中,80 °C氩气气氛中反应,获得99%收率与91%ee的手性螺烯产物3aa。
在获得上述最佳反应条件之后,作者首先考察炔基底物2的应用范围进(Table 2)。结果表明,带有供电子或吸电子基的二芳炔均能顺利参与上述反应,获得相应的手性产物3aa–3ai,收率为72-99%,ee为85-95%。同时,具有杂环取代基的炔基底物,例如噻吩基,同样以53%的收率与94%ee获得产物3aj。而且,二烷基炔底物,同样能够以94%的收率与86%ee获得产物3ak。此外,作者进一步发现,上述反应条件同样能够应用于克级规模反应,并获得良好的收率与对映选择性。
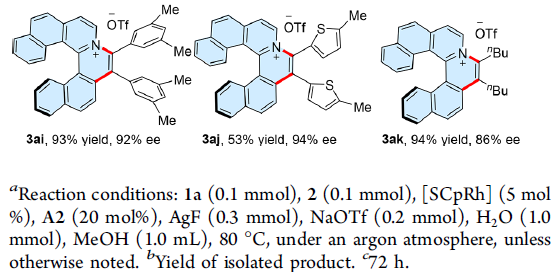
接下来,作者对1-芳基异喹啉底物1的适用范围进行进一步研究(Table 3)。实验表明,一系列带有不同取代基的1-芳基异喹啉衍生物均能良好地兼容,并以84-99%的收率与81-96%ee %获得相应的手性阳离子氮杂[6]螺旋烯产物3ba–3ja。而且,上述反应条件对于底物1k,同样具有良好的兼容性,并以90%的收率与90%ee获得手性产物3ka。同时,该反应条件同样能够用于阳离子氮杂[5]螺旋烯分子的构建,然而,要获得良好的对映选择性,需对底物进行为细致的筛选。例如,上述条件下,获得的产物3la,尽管产率良好,为88%,然而,由于未取代的[5]螺旋烯分子对映异构化能垒较低,因此,无法控制反应过程的对映选择性。随后,作者发现,在上述反应条件下,却能够以良好的产率与对映选择性获得在C-11位置具有甲基的构型稳定的阳离子氮杂[5]螺旋烯产物3ma (98% yield, 87% ee)或在C-12位置具有甲氧基的产物3na (91% yield, 93% ee)。此外,研究表明,采用外消旋的底物1o,同样可以获得96%的收率与78%ee的产物3oa。

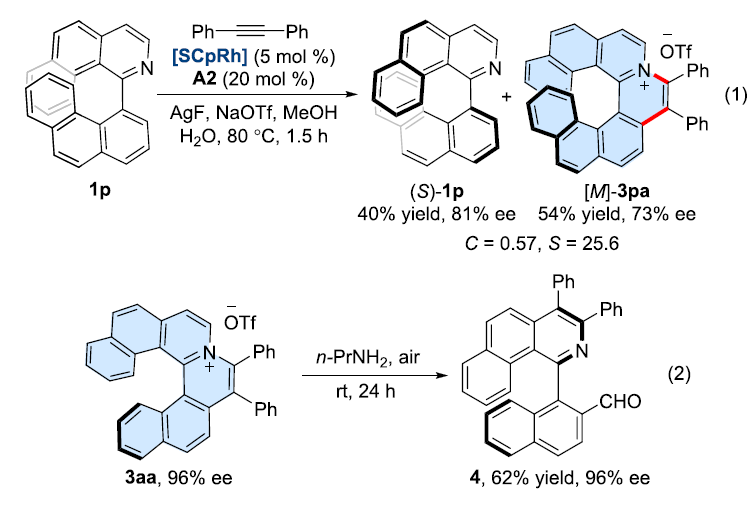
此外,该小组在底物1p中观察到动力学拆分过程,以54%的收率与73%ee获得环化产物3pa,并以40%的收率与81%ee的回收起始原料1p(eq 1)。同时,作者进一步发现,在空气以及n-PrNH2存在下,3aa反应能够十分容易地转化为轴手性化合物4,产率为62%,并且,反应过程中无对映体纯度的损失(eq2)。
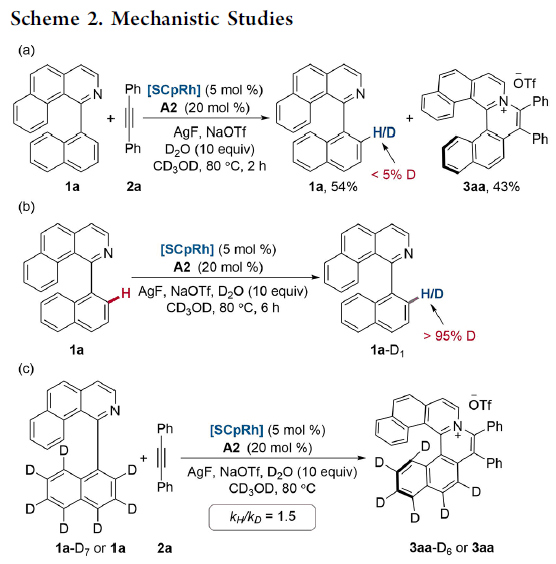
为推测上述反应的机理,作者进行一系列相关的控制实验(Scheme 2)。首先,作者发现,在1a与2a的反应体系中加入含有D2O的CD3OD时,未观察到H/D同位素置乱(H/D scrambling, Scheme 2a)。 接下来,作者在无2a存在的条件下,观察到1a能够较为容易的发生氘代,并获得>95%氘代底物1a-D1(Scheme 2b)。上述结果表明,炔基的插入速率大于铑配合物质子化的速率。接下来,作者通过氘同位素标记的相应底物,通过进行相关的动力学研究,最终确定1a与2a以及1a-D7与2a两组平行反应的kH/kD值为1.5(Scheme 2c),综上结果表明:决速步骤中可能涉及C-H键的断裂。
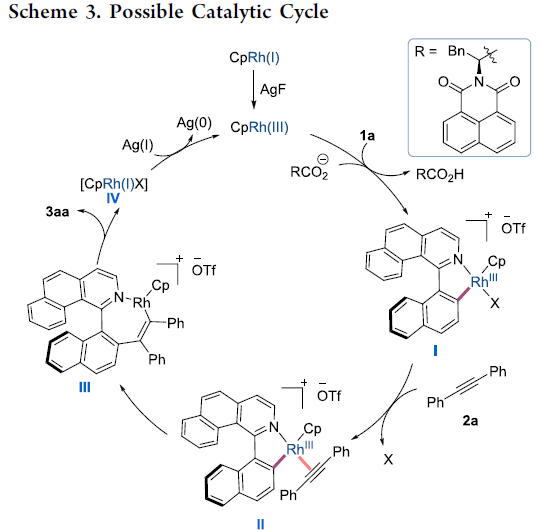
根据上述的实验与之前的文献报道[2],作者提出可能的反应机理(Scheme 3)。首先,底物1a与CpRh(III)配合物配位后,通过羧酸盐辅助的协同金属化-去质子化过程(CMD),对映选择性地断裂C-H键,获得铑环间体 (rhodacycle intermediate)I,这一过程决定产物的对映选择性。接下来,炔基底物2a与铑中心配位,并提高Rh-C键的插入,形成铑环中间体III。随后,通过III的还原消除过程,获得产物3aa与CpRh(I)配合物IV。同时,AgF的加入,能够将CpRh(I)配合物IV氧化,从而使CpRh(III)配合物再生,最终完成一次催化循环。
总结
上海有机所游书力研究员报道了一种采用手性铑(III)配合物催化的1-芳基异喹啉衍生物与炔基化合物之间的对映选择性C-H活化/环化反应方法学。同时,通过采用手性Rh(III)催化剂与手性羧酸的结合,能够以良好至极佳的收率与对映选择性获得相应的手性阳离子氮杂[5-7]螺烯类化合物。同时,反应机理研究表明,C-H键的断裂可能是上述反应过程的转化限速步骤(turnover-limiting step)。


















No comments yet.