
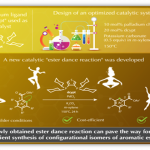
课题的提出
最近几十年,金属催化主要用于C-H键的活化,构建新的C-C键。但该催化体系在催化交叉偶联反应时极易伴随自身偶联副产物的生成[1]。至今为止,还没有关于能完全抑制自身偶联副产物生成的报道。考虑到Fe催化C-H键活化过程中会与Lewis酸作用生成有机铁中间体III[2], 东京大学Eiichi Nakamura、尚睿团队猜想若底物带有碱性基团,则两个底物可通过碱性基团发生瞬态连接形成中间体II和III的平衡态,然后,III缓慢脱去质子可生成中间体IV,紧接着,IV发生交叉偶联反应,得到产物V(Figure 1)。
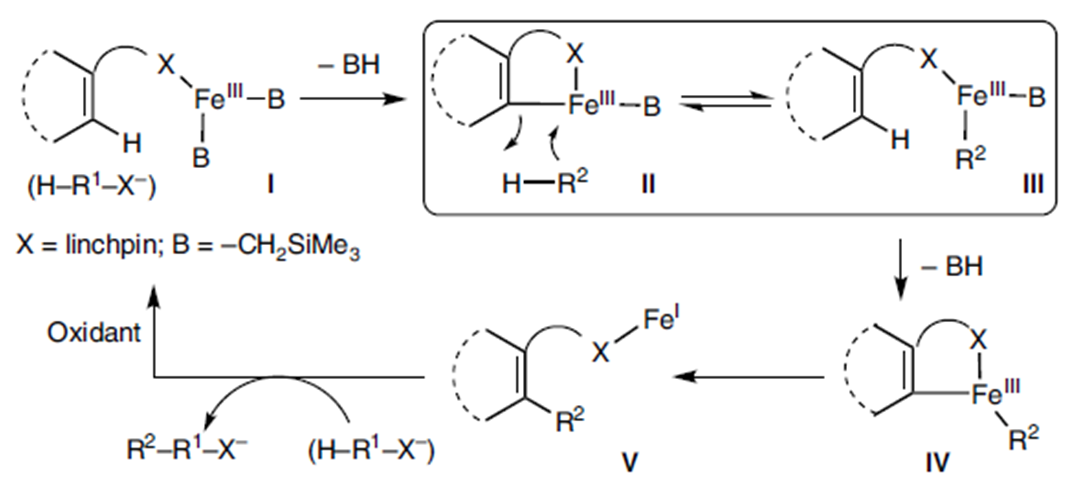
Figure 1.机理推测
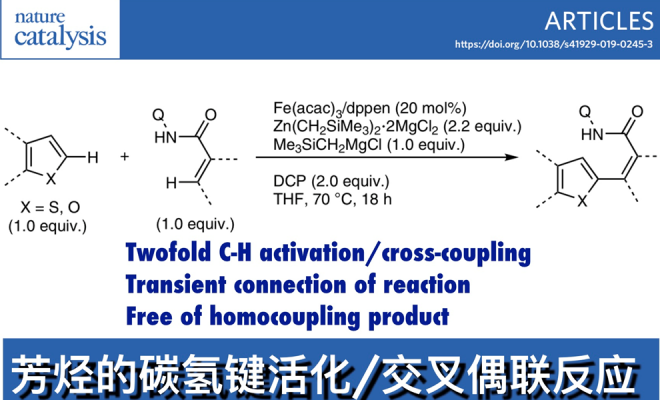
在上述背景研究的基础上,Eiichi Nakamura、尚睿团队报道了第一例Fe催化等化学当量的(杂)芳烃和(杂)芳烃或带N-(喹啉-8-基)酰胺基团的烯烃在70℃条件下发生交叉偶联反应,能以高收率得到产物且不含任何自身偶联副产物。相关研究成果发表于。
Homocoupling-free iron-catalysed twofold C–H activation/cross-couplings of aromatics via transient connection of reactants
Doba,T.;Matsubara, T.; Ilies, L.; Shang, R.;* Nakamura, E.*Nature Catal. 2019, early view
DOI: 10.1038/s41929-019-0245-3.
论文作者介绍

研究者:Eiichi Nakamura教授
研究者经历
- 1973 B.S. Faculty of Science, Tokyo Institute of Technology (Professor TeruakiMukaiyama)
- 1978 Ph.D in chemistry, Department of Chemistry, Tokyo Institute of Technology (Professor Isao Kuwajima)
- 1978-1980 Postdoctoral Research Associate, Department of Chemistry, Columbia University, New York (Professor Gilbert Stork)
- 1980-1984 Assistant Professor, Department of Chemistry, Tokyo Institute of Technology
- 1984-1993 Associate Professor, Department of Chemistry, Tokyo Institute of Technology
- 1989-1991 Adjunct Associate Professor, Department of Applied Molecular Science, Institute for Molecular Science
- 1993-1995 Professor, Department of Chemistry, Tokyo Institute of Technology
- 1995-2016 Professor, Department of Chemistry, The University of Tokyo 2016-present Molecular Technology Innovation Chair Professor, The University of Tokyo
- 2004-2010 ERATO Nakamura Functional Carbon Cluster program research director, Japan Science and Technology Agency (JST)
- 2007-2012 The University of Tokyo, Chemistry Innovation Gobal COE Program Leader
- 2008-2009 Chairman, Department of Chemistry, The University of Tokyo
研究领域
- Physical Organic Chemistry/Synthetic Chemistry/Nano-science
- Major Research Interest Physical organic chemistry directed toward creation of new reactions, new molecules and materials, and new functions. Exploration of methodologies for physical organic chemistry such as high-resolution electron microscopy.
论文概要
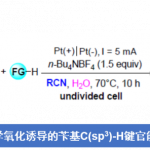
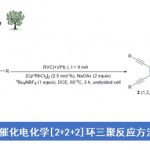
以苯并[b]噻吩和3-甲基-N-(喹啉-8-基)苯甲酰胺为模板底物,作者对反应条件进行反复筛选,确定最佳条件为(Table 1):20 mol% Fe(acac)3和共轭双膦配体dppen作为最优催化剂,2.2 equiv 有机锌试剂Zn(CH2SiMe3)2⋅2MgCl2和1 equiv Me3SiCH2MgCl为最优碱,2 equiv DCP作为温和的氧化剂,THF为最优溶剂,在70 ℃条件下反应18 h,能以97%的收率得到目标产物。
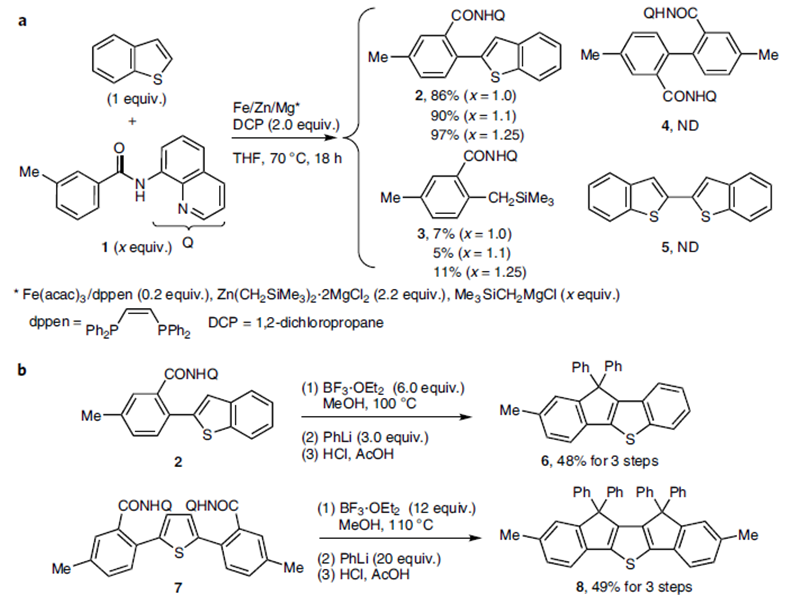
Table 1铁催化的双重C-H活化/交叉偶联及其在材料合成中的应用。
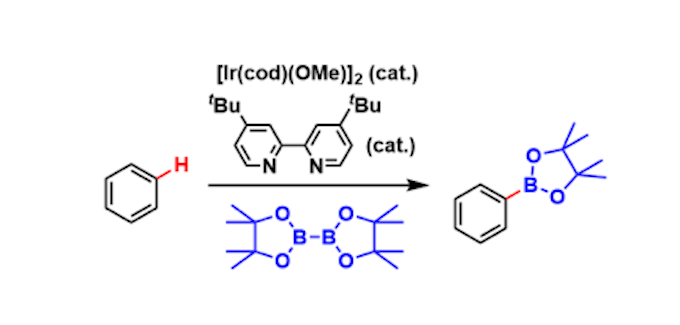
各种C-2、C-3位苯基取代的噻吩、各种苯并[b]噻吩、苯并呋喃以及带给电子的芳烃甲酰胺均能很好地适应反应条件,可以良好的收率得到相应单芳基化产物。吲哚-2-甲酰胺、噻吩-2-甲酰胺与噻吩衍生物发生偶联反应,能以较好的收率得到双杂芳烃化合物。环烯酰胺以及非环状链烯醇也能较好的适应反应条件,能以良好的收率得到Z-构型产物。1-甲基-1H-吡唑则只能以较低的收率得到相应产物。苯和卤代苯衍生物也能适应反应条件,但只能以较低的收率得到相应产物。考虑到瞬态连接的有效性,作者应用该策略能快速得到高收率的多噻吩化合物,且通过简单的步骤,这些化合物可被转化为有用的空穴传输材料、导电聚合体等结构。
紧接着,作者做了一系列氘代实验。通过该实验数据,作者推测可能的机理:酰胺阴离子A可转化为氮杂金属环B,完成第一次C-H键的活化。中间体B和C可以快速达到平衡,且该过程可逆。紧接着,C会发生构型改变转化为C′,然后–CH2SiMe3基团可促使酰胺C-H键发生不可逆的去质子化反应,形成中间体D。最后D发生交叉偶联反应得到目标产物(Figure 2)。
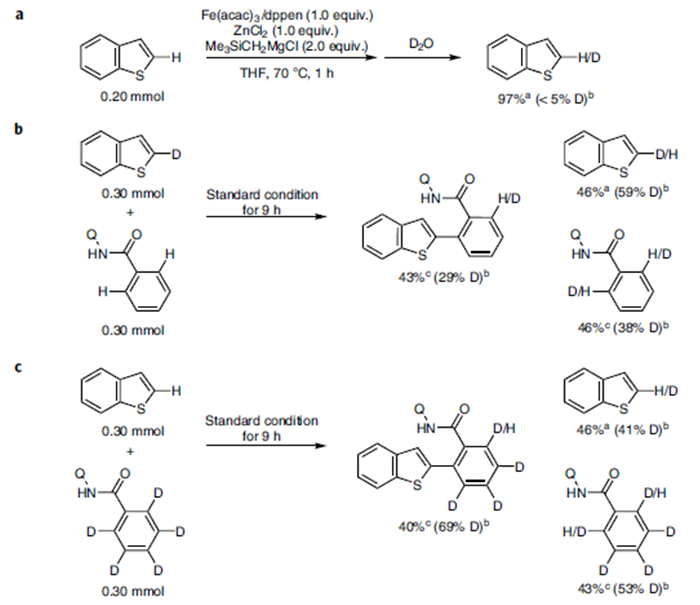

Figure 2. 氘代实验及反应机理解析
论文总结评价
Eiichi Nakamura、尚睿团队报道了第一例Fe催化等化学当量的(杂)芳烃和(杂)芳烃或带N-(喹啉-8-基)酰胺基团的烯烃发生交叉偶联反应,能以高收率得到产物且不含任何自身偶联副产物。该反应为完全抑制自身偶联副产物的生成提供了一种强有力的策略,且通过多重C-H键激活/ C-C键交叉偶联反应得到的噻吩衍生物可转化为有用的空穴传输材料、导电聚合体等结构。
参考文献
- Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787. DOI: 10.1021/acs.chemrev.6b00567
- Shang, R.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2016, 138, 10132. DOI:10.1021/jacs.6b06908
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!










No comments yet.