
作者:杉杉
导读:
近日,河南师范大学常俊标与白大昌团队在Nat. Commun.中发表论文,报道一种全新的配体调节的镍催化α-(氟烷基)苯乙烯的区域发散性氢硅化反应方法学(无需脱氟),进而成功完成了两种具有独特区域选择性的氟烷基取代硅烷分子的构建。其中,使用单齿膦配体,可生成anti-Markovnikov (β-氟烷基取代硅烷)。使用双齿膦配体,可生成更具挑战性的Markovnikov产物 (α-氟烷基取代硅烷)。同时,该反应具有原料易得、底物范围广泛、优异的区域选择性以及原子与步骤经济性等特点。机理研究表明,通过α-CF3镍中间体的配体结构控制反应的区域选择性。DFT计算揭示了一种涉及开壳单重态(open-shell singlet state)的独特机理,这对于生成复杂的四取代Markovnikov产物至关重要
Nickel-catalyzed regiodivergent hydrosilylation of α-(fluoroalkyl)styrenes without defluorination
D. Bai*, K. Zhong, L. Chang, Y. Qiao, F. Wu, G. Xu, J. Chang*.
Nat. Commun. 2024, 15, 6360. doi: 10.1038/s41467-024-50743-w.
正文:
有机氟化合物在制药、农用化学品与材料科学领域得到了广泛的应用。 其中,将三氟甲基引入有机小分子中,在药物开发与农用化学工业中具有特殊作用,可以提高其亲脂性、代谢稳定性与生物活性。然而,与已充分研究的构建C(sp2)–CF3与C(sp)–CF3键的方法相比[1],采用过渡金属催化构建C(sp3)–CF3键的方法却较少有相关的研究报道[2]。最近,诸多研究团队开发了多种利用α-(三氟甲基)苯乙烯构建高价值氟化有机化合物的方法[3]。 然而,此类反应主要集中于通过C-F键断裂构建偕-二氟烯烃及其衍生物(Fig. 1A)。另一方面,过渡金属催化烯烃的氢硅化是一种原子经济且有吸引力的制备有机硅烷的方法。然而,对于廉价金属催化末端烯烃的氢硅化的主要难题在于Markovnikov与anti-Markovnikov加成产物的区域选择性控制。2020年,Norton课题组报道了一种利用硅烷将钳形连接的Ni(II)-H物种插入α-(三氟甲基)苯乙烯中,但仅通过β-F消除合成了偕-二氟烯烃产物(Fig. 1B)[4]。这里,河南师范大学常俊标与白大昌团队报道一种全新的配体调节的镍催化α-(氟烷基)苯乙烯的区域发散性氢硅化反应方法学,进而成功完成两种具有独特区域选择性的氟烷基取代硅烷分子的构建 (Fig. 1C)。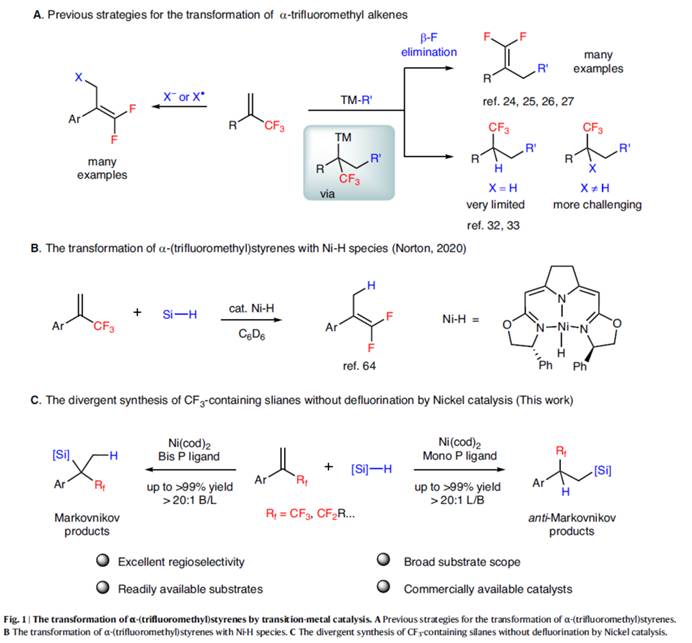
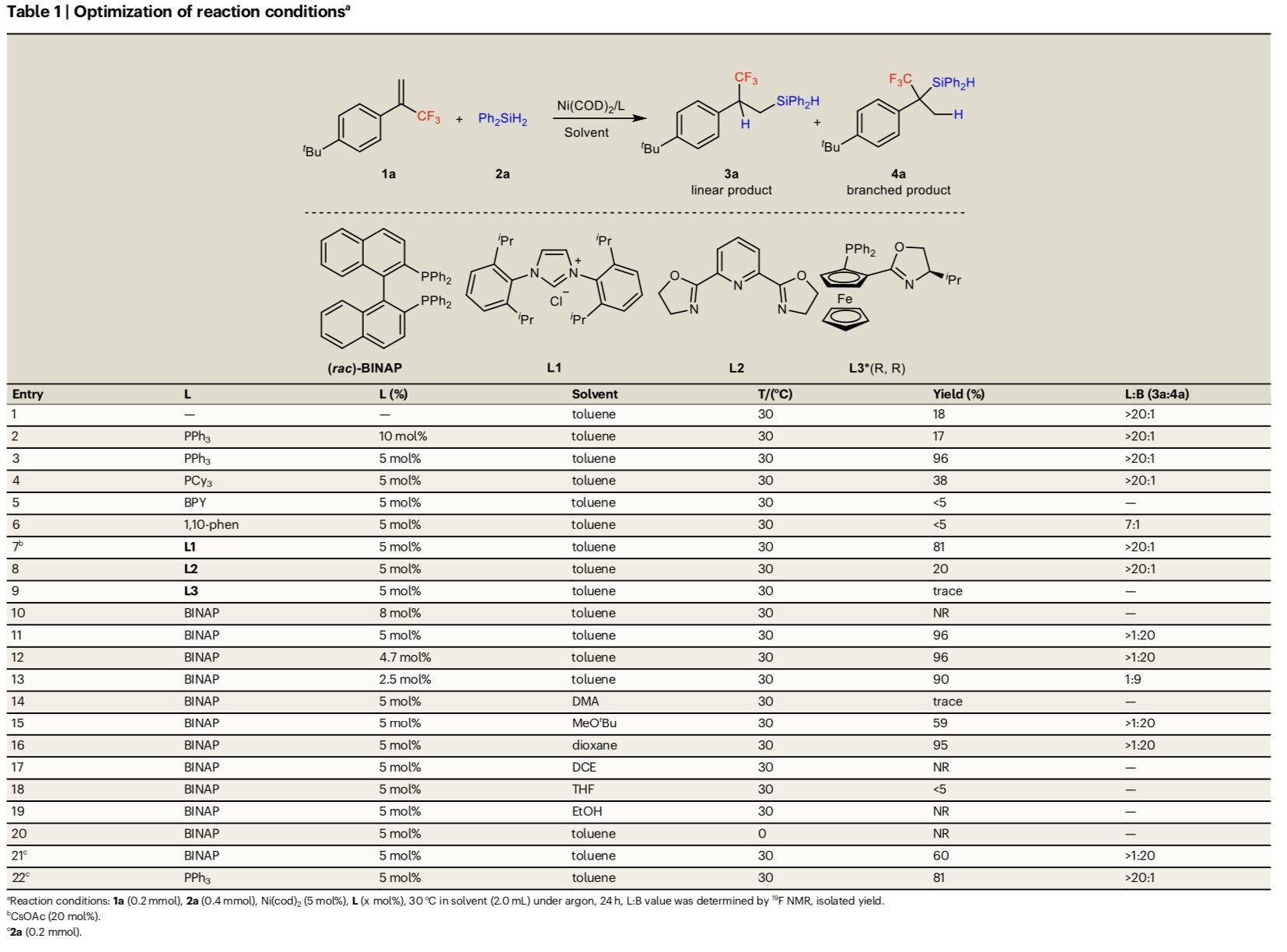
首先,作者采用苯乙烯衍生物1a与Ph2SiH2 2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用Ni(cod)2作为催化剂,PPh3作为配体,在甲苯反应溶剂中,反应温度为30 oC,最终获得96%收率的直链产物3a (L:B > 20:1)。同时,将配体改为BINAP时,可以96%的收率得到支链产物4a (L:B > 1:20)。
在上述的最佳反应条件下,作者对合成β-氟烷基取代硅烷的底物范围的进行深入研究 (Fig. 2)。首先,当底物1中的芳基上含有一系列供电子、吸电子与卤素等官能团时,均可顺利进行反应,获得相应的产物3a–3n,收率为40-99%,L:B 为7:1->20:1。其中,由于空间位阻的原因,导致产物3n的收率偏低(40%)。值得注意的是,一系列活性的基团(如烷氧羰基、胺等),均与体系兼容。然而,其他对还原条件敏感的官能团,如醛、酮与亚胺,与这种氢化硅烷化反应不相容。其次,一系列(杂)芳基取代与联芳基烯烃,也能够顺利进行反应,获得相应的产物3o–3z与3aa–3ac,收率为54-99%,L:B 为9:1->20:1。其中,对于富电子(杂)芳基取代的烯烃,使用NHC配体L1,可使反应具有优异的区域选择性,如3u–3x。此外,PhMeSiH2也是合适的底物,可以79%的收率得到anti-Markovnikov产物3ad,dr为1:1。然而,(3-(三氟甲基)丁-3-烯-1-基)苯、Ph3SiH、PhSiH3的反应均未能得到所需产物,即使在高温下也仅能回收起始原料。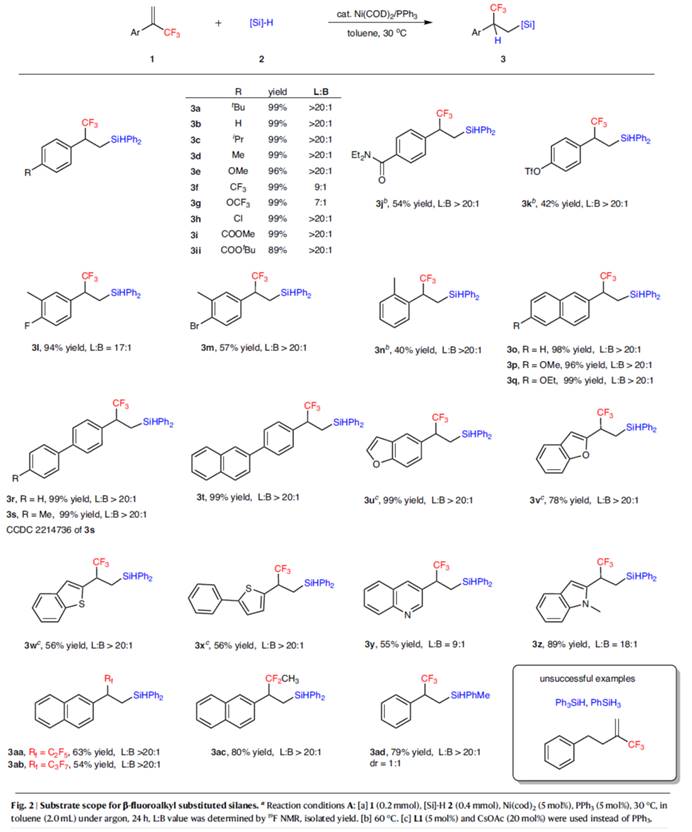
紧接着,作者对合成α-氟烷基取代硅烷的底物范围的进行深入研究(Fig. 3)。首先,当底物1中的芳基上对位含有各种官能团时,均可顺利进行反应,获得相应的产物4a–4h,收率为86-99%,B:L > 20:1。然而,对于醛、酮、亚胺与胺等还原条件敏感官能团,均与体系不相容。同时,当底物1中的芳基上含有烷氧羰基取代基,仅获得<10%收率的产物4ii。对于三氟甲磺酸酯取代的烯烃,可以38%的收率得到产物4k。当底物1中的芳基上间位含有甲基时,也能够顺利进行反应,获得相应的产物4l(收率为99%,B:L>20:1)与4m(收率为31%,B:L为9:1)。然而,当底物1中的芳基上邻位含有甲基时,由于空间效应,导致反应未能进行,如4n。其次,一系列(杂)芳基取代烯烃与联芳基取代烯烃,也是合适的底物,获得相应的产物4o–4z,收率为45-99%,B:L为5:1-> 20:1。其他氟烷基取代基的烯烃,也与体系兼容,获得相应的产物4aa(收率为46%,B:L>20:1)与4ab(收率为40%,B:L>20:1)。此外,PhMeSiH2也是合适的底物,但区域选择性与非对映选择性较差(如4ad)。然而,(3-(三氟甲基)丁-3-烯-1-基)苯、Ph3SiH与PhSiH3均未能有效的进行反应。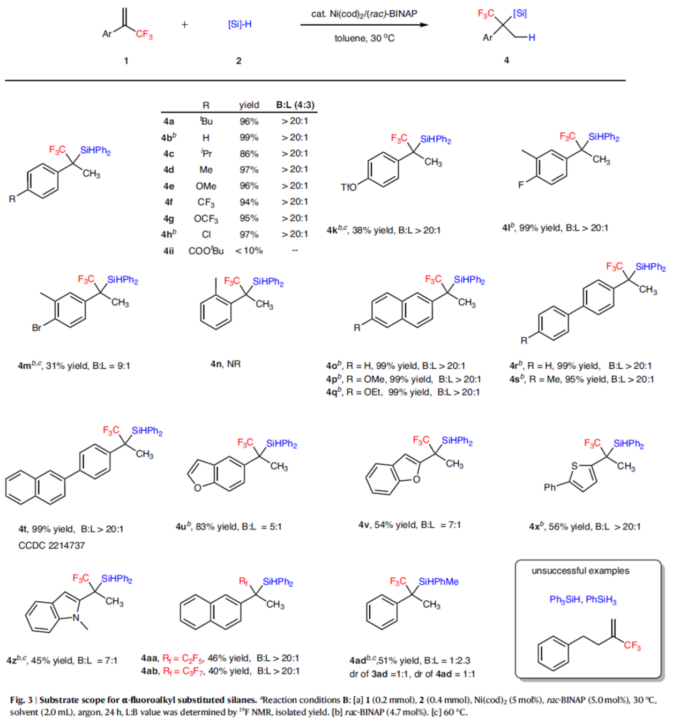 随后,作者对反应的实用性进行了研究 (Fig. 4)。首先,放大规模实验,均以99%的收率分别得到产物3r(L:B >20:1)与4r(B:L > 20:1)。其次,3r与4r可分别进行脱氟、氧化、脱氢、共轭加成等反应,分别获得相应的衍生物5–10,收率为54-89%。
随后,作者对反应的实用性进行了研究 (Fig. 4)。首先,放大规模实验,均以99%的收率分别得到产物3r(L:B >20:1)与4r(B:L > 20:1)。其次,3r与4r可分别进行脱氟、氧化、脱氢、共轭加成等反应,分别获得相应的衍生物5–10,收率为54-89%。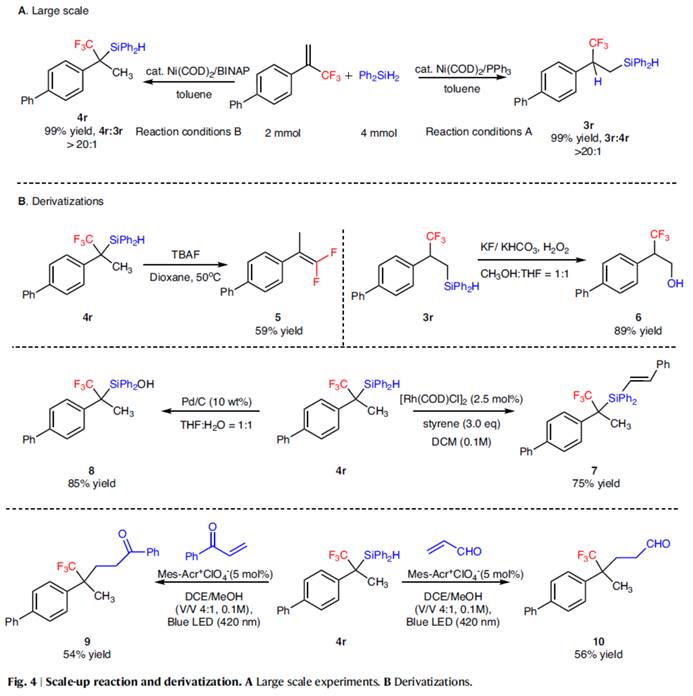
接下来,作者对上述区域发散性氢硅化过程的反应机理进行进一步研究 (Fig. 5)。首先,自由基捕获实验结果表明,反应涉及一种Chalk-Harrod反应途径,而不是自由基途径(Fig. 5A)。EPR实验也支持这一结果(见SI)。通过对末端烯烃中三氟甲基的作用研究表明,三氟甲基是一个独特的σ-吸电子基团,对于区域选择性形成Markovnikov产物4至关重要(Fig. 5B)。同时,在Conditions A下,环丙基取代的烯烃得到开环产物13。这种区域选择性与Me或iPr取代的烯烃相反,可能是由于环丙基取代基的有利β-C消除,表明NiH插入与β-H消除是可逆的。产物13的形成表明了NiH的插入途径,而非Ni-Si的插入途径。氘代实验结果表明,当PPh3用作配体时,Ni-D配合物(来自Ph2SiD2)插入烯烃1d,然后β-H消除,得到NiH配合物。而当BINAP用作配体时,在Conditions B下获得氘代产物4d-dn,硅原子上掺入93%的氘,甲基中仅发生8%的H/D交换。这一结果表明,β-H消除过程不太可能与BINAP配体一起进行(Fig. 5C)。此外,平行KIE研究结果表明,Si-H键的断裂不是决速步骤。同时,NiH插入与β-H消除过程发生在C-Si键形成之前(Fig. 5D)。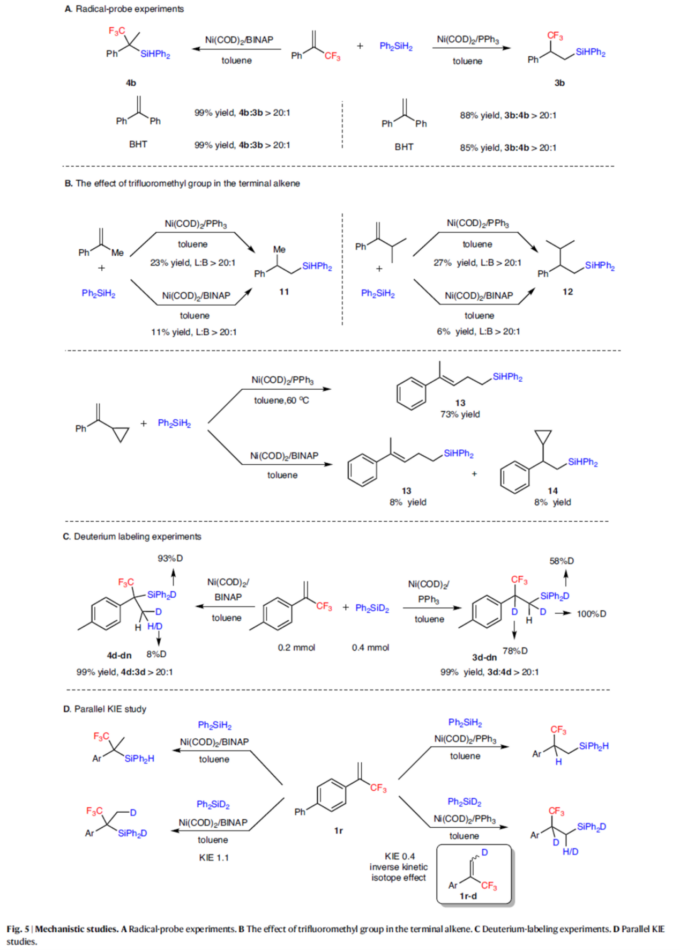
最后,作者对氢硅化反应的区域选择性起源,进行了相关的DFT计算研究(Fig. 6与Fig. 7)。在Ni/BINAP-催化合成Markovnikov产物中,Ni(0)配合物通过过渡态TS1经历[Si]-H的氧化加成,生成中间体INT2。其次,α-(三氟甲基)苯乙烯1b与NiH配合物INT2配位并经历[1,2]-插入,通过过渡态TS3生成中间体INT4。或者,NiH [2,1]-插入,通过过渡态TS2形成中间体INT3。与[2,1]-插入相比,[1,2]-插入中观察到的较低能障归因于三氟甲基的σ-吸电子效应。随后,中间体INT4中的C(sp3)–Si还原消除通过过渡态TS4发生。此外,从闭壳单线态INT4通过自旋态变化得到的开壳单线态物质INT4–oss将通过过渡态TS4–oss发生。最后,与Ph2SiH2 2a的配体交换释放Markovnikov产物3b和活性催化物种INT1,以完成催化循环。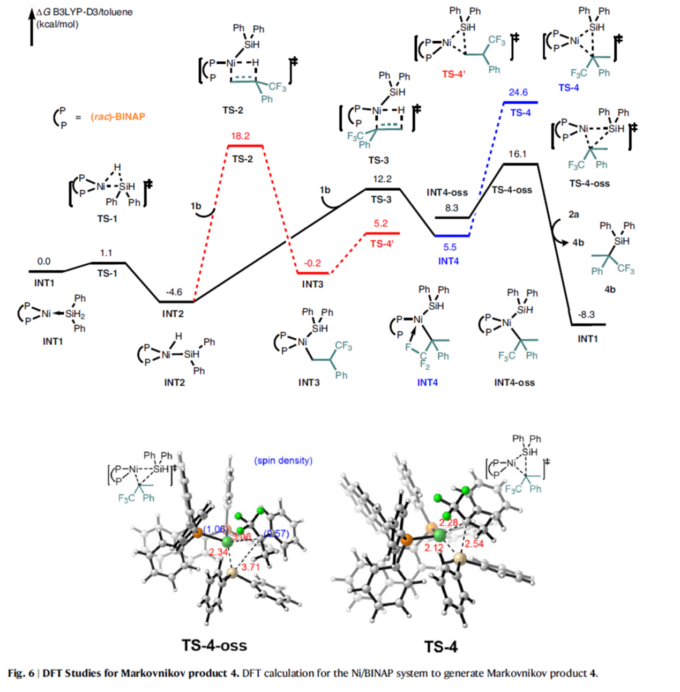
此外,在Ni/PPh3催化体系中(Fig. 7),[Si]-H通过过渡态TS5氧化加成到镍中心,生成中间体INT6。α-(三氟甲基)苯乙烯1b与镍中心配位,生成INT7,其经过[2,1]-插入,通过过渡态TS6生成中间体INT8。其次,中间体INT8中的C(sp3)–Si还原消除是通过过渡态TS7发生的。该过程最终可生成anti-Markovnikov产物3b,并通过与另一种Ph2SiH2 2a的配位再生活性催化物种INT5。或者,Ni-H经历[1,2]-插入,通过过渡态TS6’生成中间体INT9。然而,通过过渡态TS8从INT9中还原消除的C(sp3)–Si具有高能障。anti-Markovnikov产物生成的高选择性可归因于C(sp3)–Si还原消除能障的升高。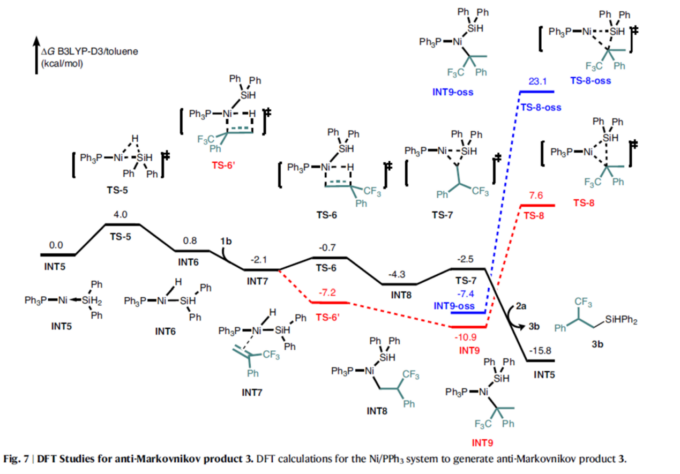
总结:
河南师范大学常俊标与白大昌团队报道一种全新的配体控制镍催化α-(氟烷基)苯乙烯的区域发散性氢硅化反应方法学,进而成功完成两种具有独特区域选择性的氟烷基取代硅烷分子的构建。机理研究表明,三氟甲基与配体的结构显著影响区域选择性。从闭壳单线态到开壳单线态的独特自旋态变化,形成了不寻常的四取代饱和Markovnikov产物。
参考文献:
- [1] L. Chu, F. Qing, J. Am. Chem. Soc. 2010, 132, 7262. doi:10.1021/ja102175w.
- [2] M. Hu, C. Ni, J. Hu, J. Am. Chem. Soc. 2012, 134, 15257. doi:10.1021/ja307058c.
- [3] F. Chen, X. Xu, L. He, G. Huang, S. Zhu, Angew. Chem. Int. Ed. 2020, 59, 5398. doi:10.1002/anie.201915840.
- [4] C. Yao, S. Wang, J. Norton, M. Hammond, J. Am. Chem. Soc. 2020, 142, 4793. doi:10.1021/jacs.9b13757.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.

















No comments yet.