
投稿作者齐藩
众所周知,芳基化反应可以通过亲电取代和过渡金属催化碳氢键官能化反应实现,尽管在过渡金属催化的官能化反应中,偶氮基团作为导向基团诱导实现了很多碳氢键官能化反应1,但是通过有机催化去实现这一转化还没有任何报道。过去几十年金属催化的碳-氢键活化在芳烃官能化方面取得了很大的发展,但是目前有机催化实现的碳氢键官能化反应很少,仅限于Nicewicz教授报道的光氧化还原有机催化剂催化的芳烃的胺化和腈化反应2。本期内容介绍的这一论文是实现有机催化的芳香亲核取代反应设计为基础,以实现轴手性化合物的不对称合成为目标,利用偶氮基团作为导向和活化基团,以手性磷酸催化为催化剂实现了偶氮苯化合物与吲哚不对称芳基化反应构建了芳基吲哚类轴手性化合物,通过不同取代的吲哚底物意外得到了邻苯胺芳基和芳基吲哚轴手性化合物,通过催化剂的优化,控制实现了这一产物的高产率高对映选择性的合成。通过改变吲哚2、3位取代基样式,可高效获得不同结构的含相邻季碳中心的手性吡咯吲哚啉产物。 这一工作通讯作者为南方科技大学谭斌副教授。
Organocatalytic asymmetric arylation of indoles enabled by azo groups.
Qi, L.-W.; Mao, J.-H.; Zhang, J.; Tan, B.
Nat. Chem. 2017, .DOI: 10.1038/NCHEM.2866
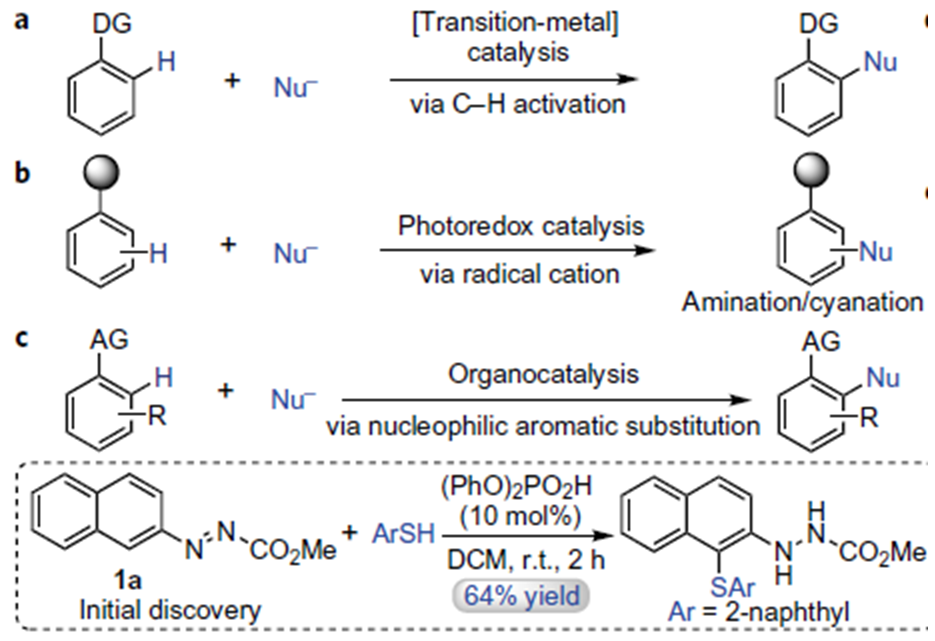
图1. 通过亲核取代反应直接实现芳基化反应
这里作者首先使用强亲核试剂萘硫醇去试探反应的可能性,发现化合物1a与萘硫醇可以有效地参与反应,并且得到目标产物产率为64%。这一反应表明布朗斯特酸催化剂可以使得芳烃具有足够的亲电性使得亲核进攻成为可能这一策略是可行的。
-
手性膦酸催化偶氮导向的吲哚亲核取代反应
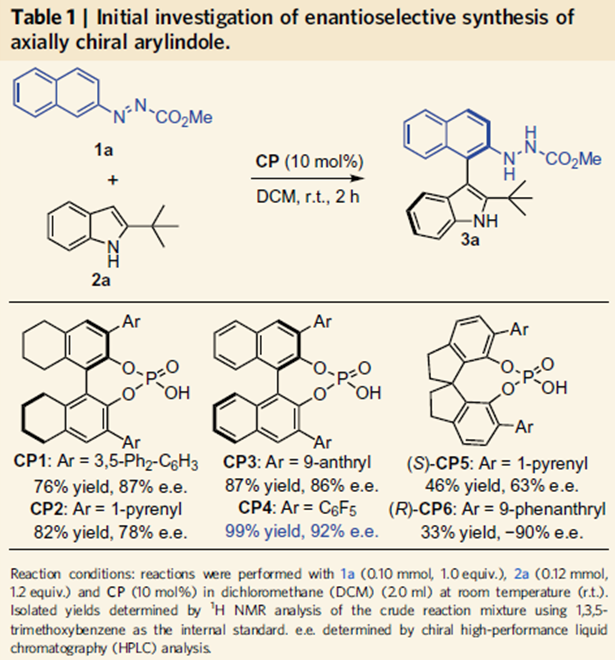
图2. 催化剂的筛选与优化
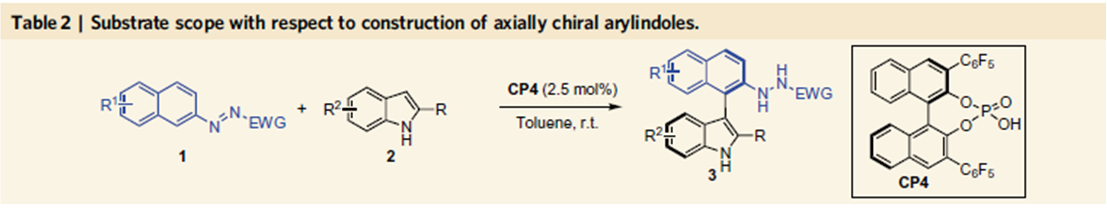
图3. 芳基吲哚类化合物合成
由于作者对轴手性联芳基化合物合成的长期兴趣,然而轴手性芳基吲哚类化合物合成还没有相关报道,因此作者选择吲哚2、3位取代的吲哚作为底物,在10 mol%手性膦酸作用下,在二氯甲烷室温下反应得到化合物3a,其中联萘酚型手性膦酸表现最好,当邻位为全氟苯取代基反应效果最好,产率为99%,对映体过量值为92%。经过溶剂,催化剂量的筛选得到反应的最优条件,其中催化剂量可以降至2.5 mol%,这时产率为95%,对映体过量值为99%。
当催化剂量降至0.05 mol%,反应依旧能够高效地进行,产率和对映选择性都能够保持。
-
酸催化吲哚的重构化反应
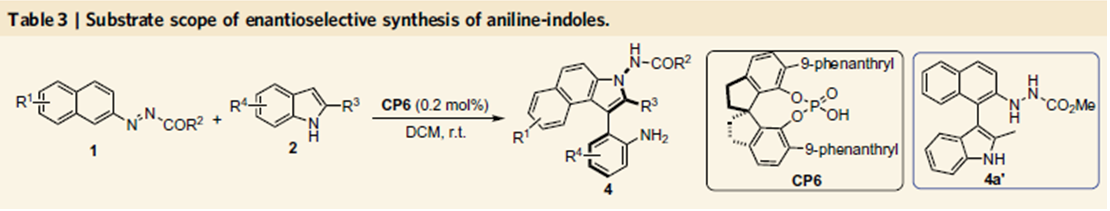
图4. 苯胺吲哚类化合物的合成
在Table 1中3系列化合物的拓展中,作者偶然发现了一种意外的产物,当2-甲基吲哚为底物时,得到的产物为吲哚去芳基化重排的产物,而在Table 1中2位为叔丁基取代的吲哚,由于位阻的影响,因此在Table 1中没有出现这种产物。作者通过催化剂筛选发现,CP6反应效果最好,产率为87%,对映体过量值为99%。后面的底物拓展表明,2位为异丙基取代的吲哚时,反应也可以很好地进行,这表明反应可以适用于2位具有一定位阻的吲哚。
-
手性吡咯吲哚啉的合成
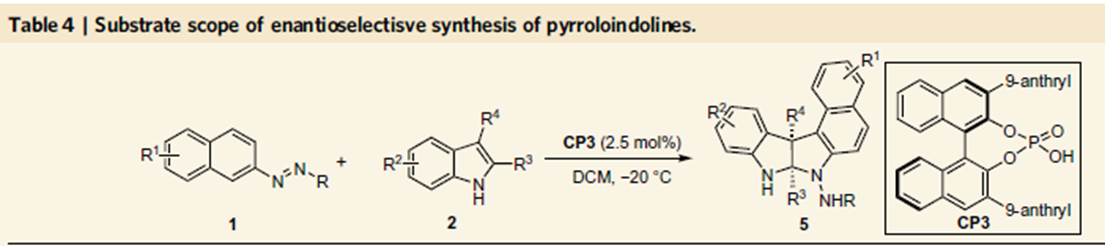
图5. 手性吡咯吲哚啉化合物的合成
前两个反应的结果这一有机催化芳基化反应的可行性,小编认为如果可以控制吡咯的去芳基化,这时构建手性吡咯吲哚啉产物是自然而然的事情,不过这时会出现两个相邻的季碳手性中心,这里的难题在于如何有效地实现这一反应的立体化学控制。反应条件的探索中发现,反应在常温下,反应的产率和立体化学控制均没有达到最理想的水平,尤其是对映选择性控制上,反应在0℃时,产率为95%,ee值为97%。当催化剂量由10%将至2.5%时,-20℃时,ee值为99%。底物拓展表明,吲哚底物并不仅限于2,3均为甲基取代的吲哚,3位为烯丙基取代,2位为乙基取代也适用,并环的底物如四氢咔唑并吲哚,四氢环戊二烯并吲哚也能够适用于这一反应,取得了很好的反应效果。放大实验表明,以上三个反应均可以实现克级制备,产率和对映选择性不受影响。
-
可能的反应机理
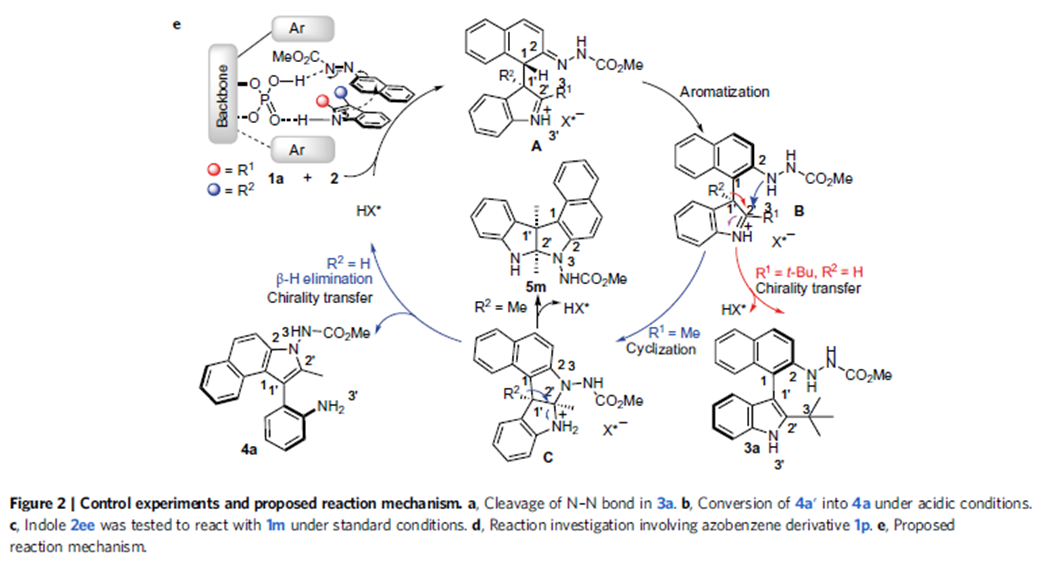
图6. 可能的反应机理
在这一反应机理中,手性膦酸充当双功能催化剂,或者小编认为更像是一个手性口袋,通过双氢键作用底物,在这一手性口袋中,吲哚对映选择性进攻偶氮芳烃的α位,芳构化脱去质子得到轴手性化合物3。中间体B在酸作用下发生分子内环化,这一环化过程相当于苯胺对亚胺的加成得到中间体C,经β氢消除原来的吲哚去芳构化,在引导基团邻位吲哚环得以重新构建得到4a。中间体C也可以直接脱去质子得到手性吡咯吲哚啉化合物5。
-
结论
作者发现偶氮基不仅可以有效激发芳烃亲核进攻,而且可以有效地引导芳烃的亲核取代反应,这一反应可以发展高效的有机催化的对映选择性吲哚的芳基化反应。这一反应的优势如下,1、利用偶氮苯衍生物为亲电试剂,实现了有机催化的亲核取代反应,这一反应也实现了有机催化地芳烃碳氢键的不对称芳基化反应。2、 在这一反应中,偶氮基作为引导基团和活化基团。3、 轴手性芳基吲哚类化合物,苯胺吲哚类化合物、吡咯吲哚啉化合物可以高产率、高对映选择性地被合成,克级试验中产率和对映选择性不受影响。4、反应温和,反应条件简单,催化量低,在轴手性芳基吲哚类化合物的合成中催化量降至0.05 mol%,反应仍然可以高效进行。
参考文献:
- (a) Dick, A. R.; Hull, K. L.; Sanford, M. S. A Highly Selective Catalytic Method for the Oxidative Functionalization of C−H Bonds. Am. Chem. Soc. 2004,126 (8), 2300-2301. DOI:10.1021/ja031543m. (b) Li, H.; Li, P.; Wang, L. Direct access to acylatedazobenzenes via Pd-catalyzed C–H functionalization and further transformation into an indazole backbone. Org. Lett. 2013, 15, 620–623. DOI: 10.1021/ol303434n.(c) Wangweerawong, A.; Bergman, R. G.; Ellman, J. A. Asymmetric synthesis ofα-branched amines via Rh(III)-catalyzed C–H bond functionalization. J. Am. Chem. Soc.2014, 136, 8520–8523. DOI: 10.1021/ja5033452.
- (a) Romero, N. A.; Margrey, K. A.; Tay, N. E.; Nicewicz, D. A. Site-selective arene C–H amination via photoredox catalysis. Science. 2015. 349, 1326– DOI: 10.1126/science.aac9895. (b) McManus, J. B.; Nicewicz, D. A. Direct C–H cyanation of arenes via organic photoredox catalysis. J. Am. Chem. Soc.2017, 139, 2880–2883. DOI:10.1021/jacs.6b12708.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!





















No comments yet.