
本文作者:杉杉
导读
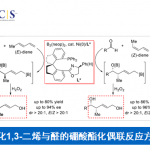
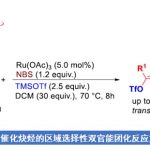
烷基卤化物(外消旋)与杂芳烃C(sp2)-H键直接对映发散性交叉偶联(enantioconvergent)的发展受到阻碍,由于在高温下使用碱会发生消旋化(racemization)。近日,南方科技大学刘心元课题组在Angew. Chem. Int. Ed.上发表论文,通过铜(I)/金鸡纳生物碱(cinchona-alkaloid)衍生的N,N,P-配体的催化体系,该体系能够在温和条件下与消旋的烷基溴化物进行氧化加成。因此,已实现烷基卤化物与唑C(sp2)-H键的高对映选择性交叉偶联,从而获得多种具有价值的α-手性烷基唑化合物,如1,3,4-恶二唑、恶唑、苯并[d]恶唑、1,3,4-三唑等。此外,反应机理涉及唑C(sp2)-H键的去质子化和烷基自由基参与的过程。
Copper-Catalyzed Enantioconvergent Cross-Coupling of Racemic Alkyl Bromides with Azole C(sp2)-H Bonds
Chunlan Song, Xiao-Long Su, Liu Ye, Ji-Jun Chen, Xiao-Dong Liu, Sheng-Peng Jiang, Fu-Li Wang, Lin Liu, Chang-Jiang Yang, Xiao-Yong Chang, Zhong-Liang Li, Qiang-Shuai Gu, and Xin-Yuan Liu
Angew. Chem. Int. Ed. ASAP, DOI: 10.1002/anie.202009527
正文
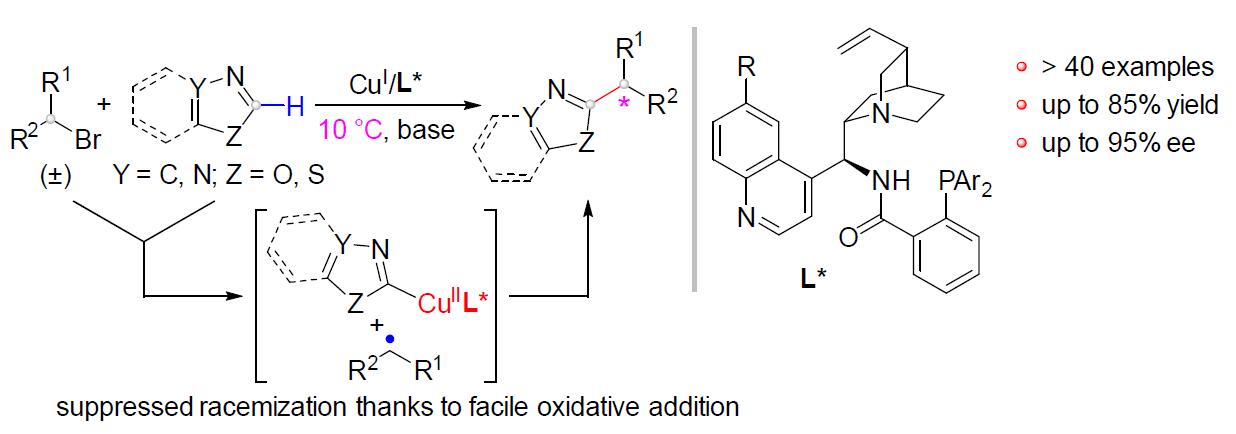
α-手性烷基杂芳烃广泛存在于生物活性的小分子、天然产物和药物中。在过去二十年中,过渡金属催化烷基卤化物的对映发散性自由基C(sp3)-C交叉偶联受到广泛的关注。通过多种预官能化的(杂)芳基试剂与烷基卤化物对映发散性(杂)芳基化偶联,已经成为构建α-手性烷基(杂)芳烃的有效方法(Scheme 1A)。因此,由Fu,Reisman等[1-4]提出的策略,使用手性过渡金属(如Ni,Co和Fe)催化剂,可将消旋的烷基亲电体转化为前手性烷基。尽管该方法已取得一定的结果,但从底物的可用性、操作简便性以及C-H功能化的高原子经济性角度考虑,直接使用杂芳烃代替这些预官能化的杂芳基试剂更为有效。然而,在烷基亲电体与杂芳烃C(sp2)-H的偶联反应中,不可避免地需在高温下(80-160 ℃)使用碱,从而易发生消旋化(Scheme 1B),尤其是缺电子的杂芳烃。
基于对烷基自由基的不对称反应的研究,本课题组最近发现,手性生物碱衍生的多齿N,N,P-配体可增强铜催化剂的还原能力[5]。因此,所得的手性铜催化剂易将卤代烷还原为烷基自由基。鉴于上述消旋化问题以及唑在药物开发中的重要性,作者设想, Cu(I)/ N,N,P-配体催化剂可能会在温和条件下促进所需的卤代烷烃与唑C(sp2)-H键的对映体偶联反应。然而存在以下挑战,(1)有效抑制杂环或卤代烷烃的自偶联;(2)对烷基自由基的杂芳基对映选择性控制。如果成功的话,这种方法不仅可作为先前报道的对映选择性杂芳烃C(sp2)-H烷基化的补充 [6],而且可直接获得对映体富集的α-手性烷基化唑类化合物。

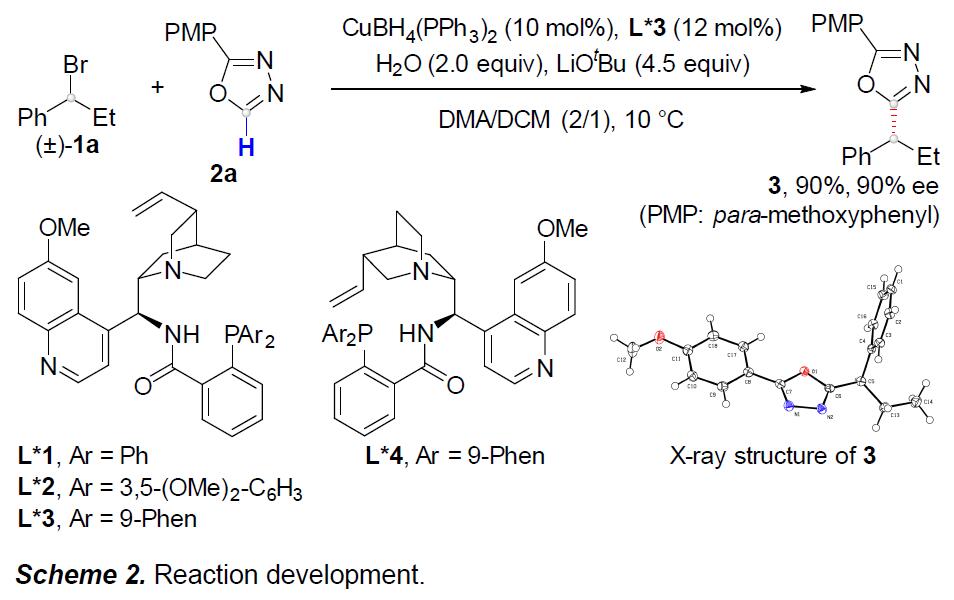
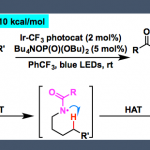
首先,作者以(±)-1a和2a作为模型底物,进行了相关偶联反应条件的筛选(Scheme 2)。反应的最佳条件为,使用(±)-1a (2 eq),2a (1 eq),CuBH4(PPh3)2 (10 mol%),L*3 (12 mol%),H2O (2 equiv), LiOtBu (4.5 eq),使用DMA/DCM (2/1, 0.60 mL)的混合溶剂,可在10 °C反应温度与氩气保护条件下,获得90%收率和90% ee.的产物 3。
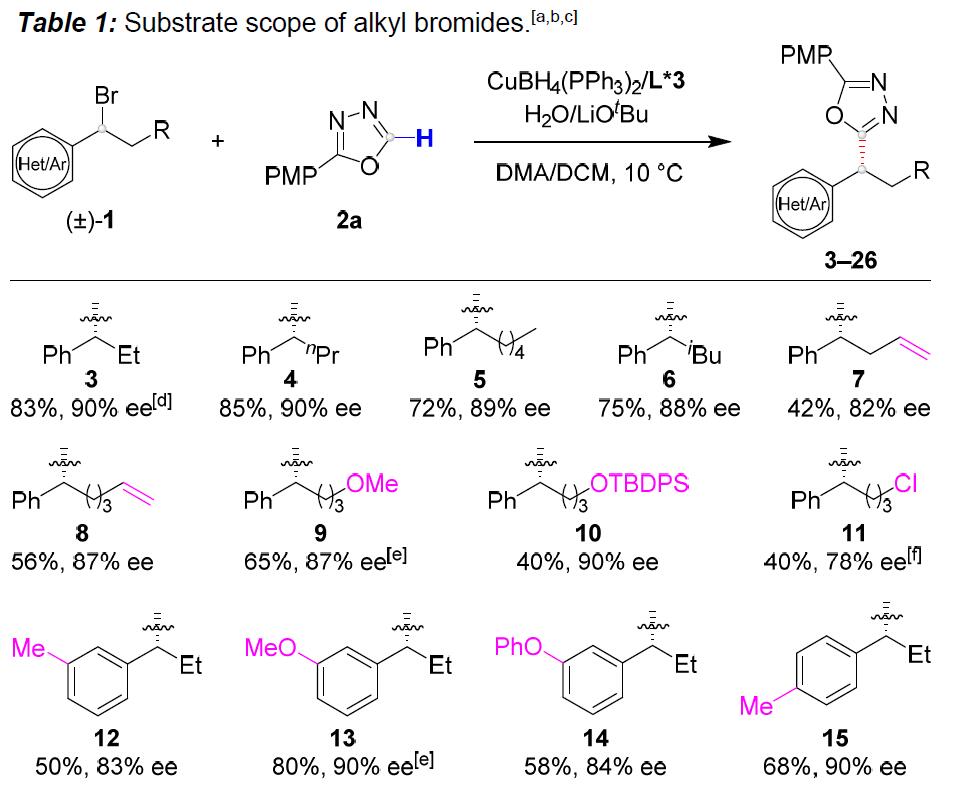
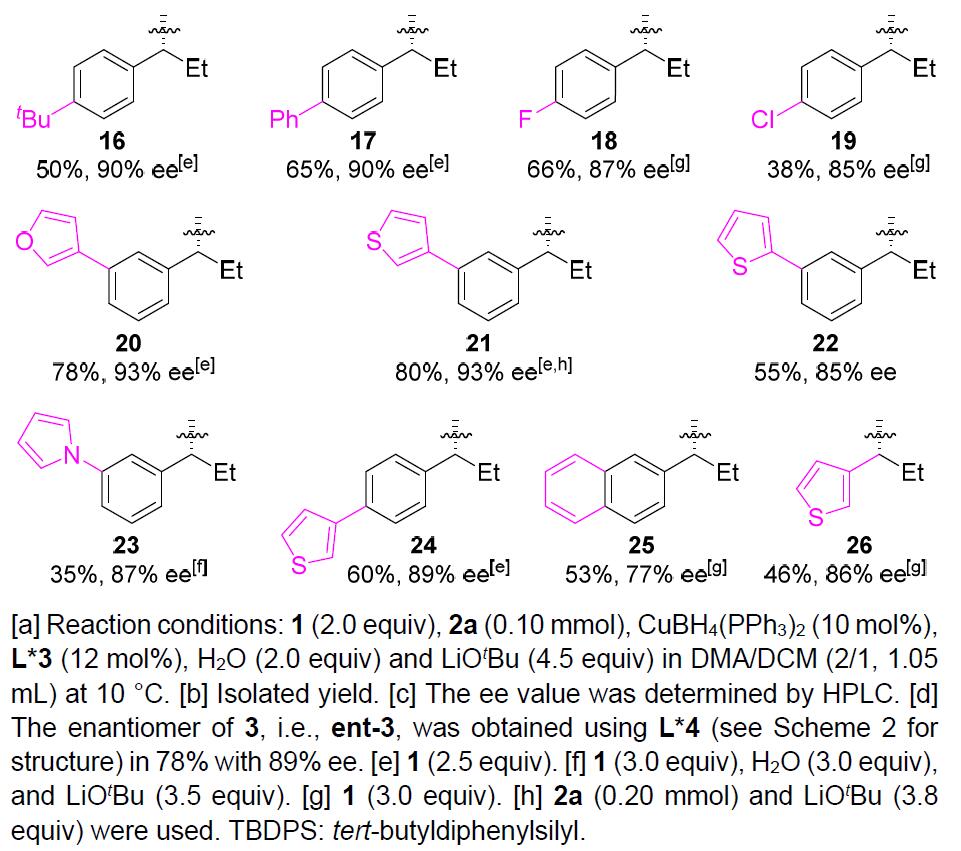
在获得上述最佳反应条件后,作者开始对烷基溴底物1进行了扩展(Table 1)。反应结果表明,具有直/支链的脂肪族苄基溴均为合适的底物(3–6),而烷基侧链上的取代基,如末端烯烃(7和8),醚(9),甲硅烷基保护的醇(10)和伯氯(11)以及苄基溴的苯环上具有给电子或吸电子的各种官能团(12–19)时,均与体系兼容。同时,杂芳基取代基如呋喃基(20)、噻吩基(21、22和24)和吡咯基(23)也适用于该反应。此外,双环2-萘基(25)和杂环3-硫代苯基取代的烷基溴(26)也是可行的底物。
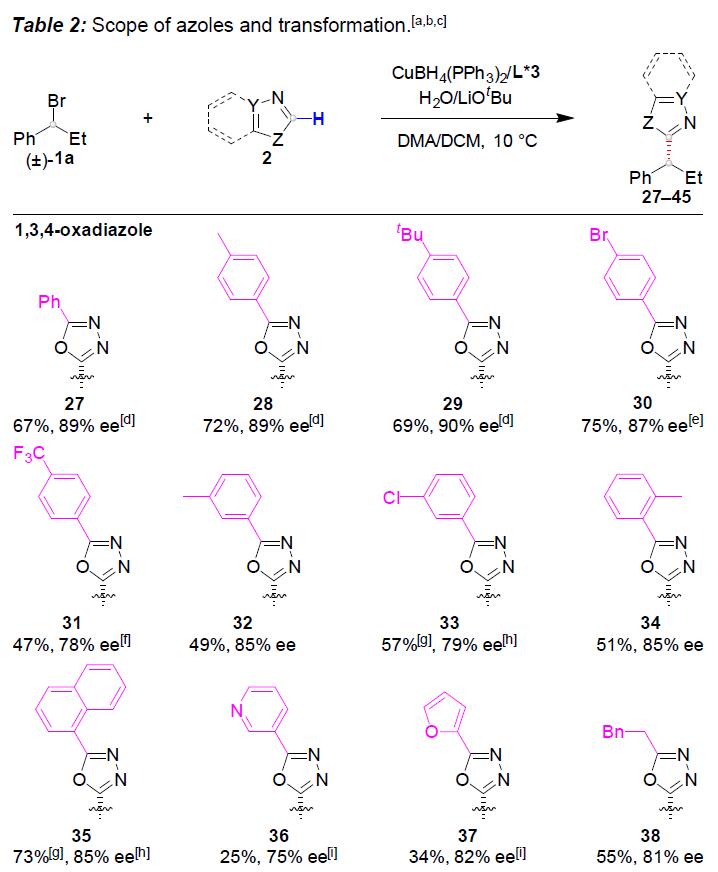
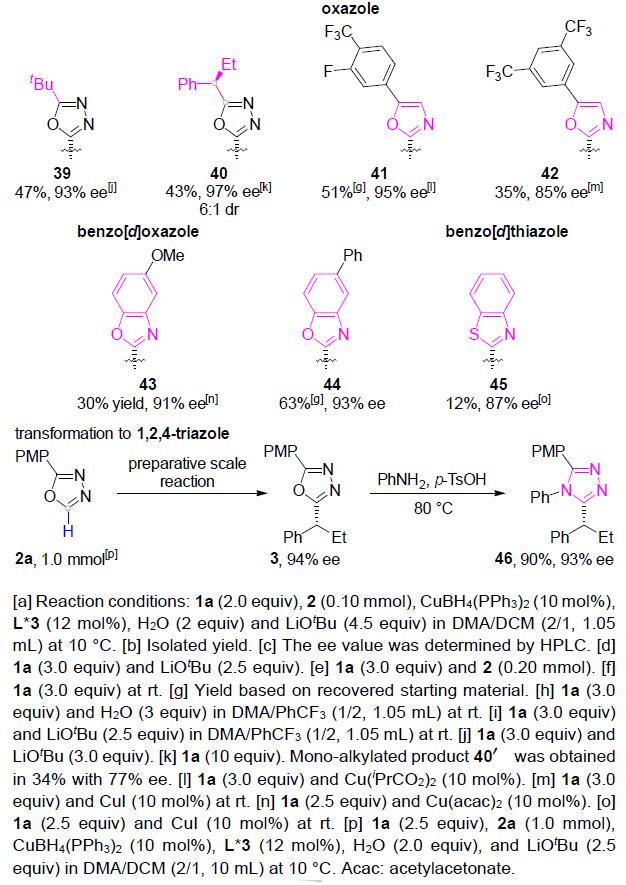
随后,作者对唑底物2进行了相关的扩展(Table 2)。各种2-苯基取代的1,3,4-恶二唑均可顺利进行烷基化反应(27-34)。带有萘基(35)、吡啶基(36)和呋喃基(37)取代基的1,3,4-恶二唑,同样与体系兼容。值得注意的是,具用烷基取代(杂)芳基取代基以中等收率和良好至优异的ee获得产物38和39。有趣的是,未取代的1,3,4-恶二唑以高对映选择性和中等非对映选择性获得双烷基化产物40。此外,恶唑、苯并[d]恶唑、苯并[d]噻唑也可作为杂芳基化试剂,以中等收率和出色的对映选择性获得产物41–45。然而,对于其他的唑类和氰基噻吩未能取得好的结果。值得注意的是,对于46的放大,同样获得预期的效果。此外,产物中含有的卤化物(11、19、30和33),可进一步衍生化,从而合成更为复杂的产物。
为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 3)。通过温度对照发现,在40℃时仅发生少量消旋,50℃时对映选择性大大降低,60℃时烷基化产物明显消旋。从而表明,该配体在低温下易于氧化加成以抑制产物消旋化作用。同时,在有或无铜时,底物2a中C5-H与过量D2O很容易进行D/H交换,表明C5-H的酸性足以直接去质子化而不需要铜。随后,使用TEMPO时,获得产物47,并未形成产物3(Scheme 3A)。此外, (±)-1b在标准条件下进行串联5-exo-trig环化和C(sp3)-C(sp2)偶联获得48和49(Scheme 3B)。因此,这些结果表明烷基自由基参与反应。此外,使用2b进行自由基钟(radical clock)实验,仅获得具有完整环丙烷环的烷基化产物50(Scheme 3C)。因此,在标准条件下,不太可能将烷基直接加到唑环上。
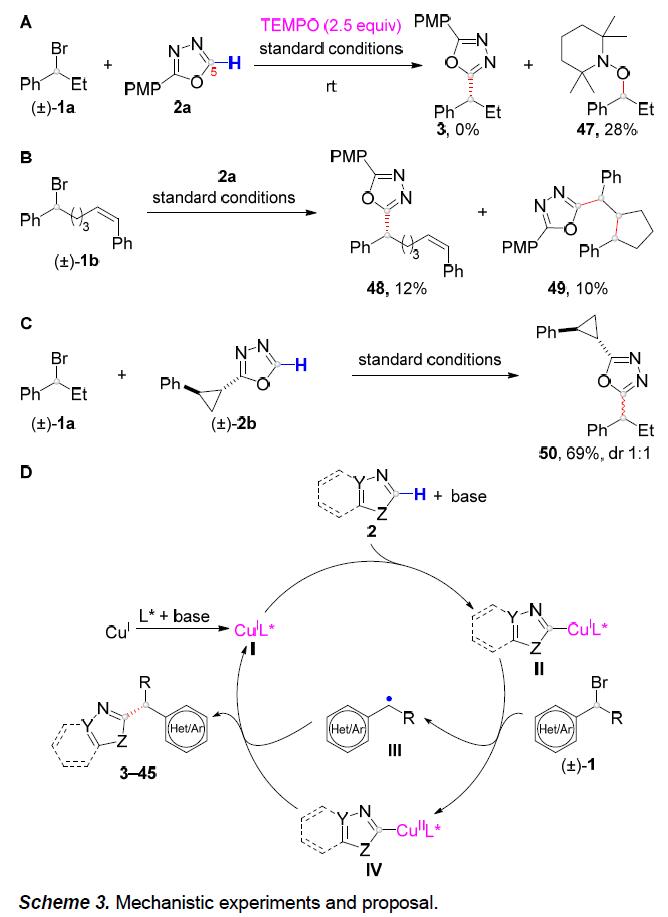
根据上述实验结果以及前期工作的总结[7-8],作者提出了一种可能的反应机理(Scheme 3D)。首先,在碱性条件下,CuIL*配合物I与唑2反应,生成中间体II。紧接着,该中间体II将烷基溴(±)-1还原为烷基自由基III,同时自身被氧化形成CuII配合物IV。最后,C(sp3)- C(sp2)偶联可能通过形成CuIII中间体及其随后的还原消除而发生。
总结
南方科技大学刘心元课题组报道了使用Cu(I)/金鸡纳生物碱衍生的N,N,P-配体的催化体系,实现烷基卤化物与唑C(sp2)-H键的高对映选择性交叉偶联,从而获得多种具有价值的α-手性烷基唑化合物,如1,3,4-恶二唑、恶唑、苯并[d]恶唑、1,3,4-三唑等。值得注意的是,降低反应温度的是N,N,P-配体提高铜的还原能力的关键,从而有效地抑制了外消旋化以及自偶联的问题。此外,反应机理涉及唑C(sp2)-H键的去质子化和烷基自由基参与的过程。
参考文献
- [1] a) J. Choi, G. C. Fu, Science 2017, 356, eaaf7230; b) G. C. Fu, ACS Cent. Sci. 2017, 3, 692; c) A. H. Cherney, N. T. Kadunce, S. E. Reisman, Chem. Rev. 2015, 115, 9587.
- [2] For selected recent examples using Ni catalysis: a) H. Yin, G. C. Fu, J. Am. Chem. Soc. 2019, 141, 15433; b) A. Varenikov, M. Gandelman, Nat. Commun. 2018, 9, 3566; for a selected example using Co catalysis: c) J. Mao, F. Liu, M. Wang, L. Wu, B. Zheng, S. Liu, J. Zhong, Q. Bian, P. J. Walsh, J. Am. Chem. Soc. 2014, 136, 17662; for a selected example using Fe catalysis: d) M. Jin, L. Adak, M. Nakamura, J. Am. Chem. Soc. 2015, 137, 7128.
- [3] For selected representative enantioconvergent reductive cross-coupling examples: a) H. Guan, Q. Zhang, P. J. Walsh, J. Mao, Angew. Chem. Int. Ed. 2020, 59, 5172; Angew. Chem. 2020, 132, 5210; b) K. E. Poremba, N. T. Kadunce, N. Suzuki, A. H. Cherney, S. E. Reisman, J. Am. Chem. Soc. 2017, 139, 5684; c) N. T. Kadunce, S. E. Reisman, J. Am. Chem. Soc. 2015, 137, 10480.
- [4] For selected representative enantioconvergent cross-coupling examples using alkyl radical precursors other than alkyl halides: a) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu, D. W. C. MacMillan, J. Am. Chem. Soc. 2016, 138, 1832; b) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander, M. C. Kozlowski, J. Am. Chem. Soc. 2015, 137, 4896.
- [5] a) X.-Y. Dong, Y.-F. Zhang, C.-L. Ma, Q.-S. Gu, F.-L. Wang, Z.-L. Li, S.-P. Jiang, X.-Y. Liu, Nat. Chem. 2019, 11, 1158; b) X.-Y. Dong, J.-T. Cheng, Y.-F. Zhang, Z.-L. Li, T.-Y. Zhan, J.-J. Chen, F.-L. Wang, N.-Y. Yang, L. Ye, Q.-S. Gu, X.-Y. Liu, J. Am. Chem. Soc. 2020, 142, 9501; for an elegant work of ligand design for copper-catalyzed radical enantioconvergent reaction: c) H. Iwamoto, K. Endo, Y. Ozawa, Y. Watanabe, K. Kubota, T. Imamoto, H. Ito, Angew. Chem. Int. Ed. 2019, 58, 11112; Angew. Chem. 2019, 131, 11229.
- [6] For selected reviews on enantioselective Friedel-Crafts alkylation of heteroarenes, see: a) M. M. Heravi, V. Zadsirjan, B. Masoumi, M. Heydari, J. Organomet. Chem. 2019, 879, 78; b) Y. Zhang, X. Liu, L. Lin, X. Feng, Prog. Chem. 2018, 30, 491; c) V. Terrasson, R. Marcia de Figueiredo, J. M. Campagne, Eur. J. Org. Chem. 2010, 2010, 2635; for selected enantioselective alkylation of heteroarenes using other strategies: d) R. S. J. Proctor, H. J. Davis, R. J. Phipps, Science 2018, 360, 419; e) X. Liu, Y. Liu, G. Chai, B. Qiao, X. Zhao, Z. Jiang, Org. Lett. 2018, 20, 6298; f) X. Bao, Q. Wang, J. Zhu, Angew. Chem. Int. Ed. 2017, 56, 9577; Angew. Chem. 2017, 129, 9705; g) J. Loup, D. Zell, J. C. A. Oliveira, H. Keil, D. Stalke, L. Ackermann, Angew. Chem. Int. Ed. 2017, 56, 14197; Angew. Chem. 2017, 129, 14385; h) M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori, V. K. Aggarwal, J. Am. Chem. Soc. 2016, 138, 9521; i) B. M. Trost, L. Debien, Chem. Sci. 2016, 7, 4985; j) H. Ohmiya, H. Zhang, S. Shibata, A. Harada, M. Sawamura, Angew. Chem. Int. Ed. 2016, 55, 4777; Angew. Chem. 2016, 128, 4855; k) C. M. Filloux, T. Rovis, J. Am. Chem. Soc. 2015, 137, 508; l) P.-S. Lee, N. Yoshikai, Org. Lett. 2015, 17, 22; m) J. Llaveria, D. Leonori, V. K. Aggarwal, J. Am. Chem. Soc. 2015, 137, 10958; n) G. Song, W. W. N. O, Z. Hou, J. Am. Chem. Soc. 2014, 136, 12209; o) C. S. Sevov, J. F. Hartwig, J. Am. Chem. Soc. 2013, 135, 2116.
- [7] For selected summaries: a) Q.-S. Gu, Z.-L. Li, X.-Y. Liu, Acc. Chem. Res. 2020, 53, 170; b) Z.-L. Li, G.-C. Fang, Q.-S. Gu, X.-Y. Liu, Chem. Soc. Rev. 2020, 49, 32; for selected examples: c) C.-J. Yang, C. Zhang, Q.-S. Gu, J.-H. Fang, X.-L. Su, L. Ye, Y. Sun, Y. Tian, Z.-L. Li, X.-Y. Liu, Nat. Catal. 2020, 3, 539; d) X.-T. Li, L. Lv, T. Wang, Q.-S. Gu, G.-X. Xu, Z.-L. Li, L. Ye, X. Zhang, G.-J. Cheng, X.-Y. Liu, Chem 2020, 6, 1692; e) Y.-F. Cheng, J.-R. Liu, Q.-S. Gu, Z.-L. Yu, J. Wang, Z.-L. Li, J.-Q. Bian, H.-T. Wen, X.-J. Wang, X. Hong, X.-Y. Liu, Nat. Catal. 2020, 3, 401; f) L. Ye, Y. Tian, X. Meng, Q.-S. Gu, X.-Y. Liu, Angew. Chem. Int. Ed. 2020, 59, 1129; Angew. Chem. 2020, 132, 1145; g) J.-S. Lin, X.-Y. Dong, T.-T. Li, N.-C. Jiang, B. Tan, X.-Y. Liu, J. Am. Chem. Soc. 2016, 138, 9357.
- [8] a) X.-Y. Dong, Y.-F. Zhang, C.-L. Ma, Q.-S. Gu, F.-L. Wang, Z.-L. Li, S.-P. Jiang, X.-Y. Liu, Nat. Chem. 2019, 11, 1158; b) X.-Y. Dong, J.-T. Cheng, Y.-F. Zhang, Z.-L. Li, T.-Y. Zhan, J.-J. Chen, F.-L. Wang, N.-Y. Yang, L. Ye, Q.-S. Gu, X.-Y. Liu, J. Am. Chem. Soc. 2020, 142, 9501.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!


















No comments yet.