
作者:杉杉
导读:
近期,河南师范大学的常俊标与白大昌等人在Nat. Commun.中发表论文,报道一种全新的无金属催化bicyclo[1.1.0]butanes (BCBs)与缺电子烯烃的极性反转Alder-ene反应方法学,进而成功完成一系列官能团化的环丁烯与共轭二烯分子的构建。
Umpolung reactivity of strained C–C σ-bonds without transition-metal catalysis
D. Bai, X. Guo, X. Wang, W. Xu, R. Cheng, D. Wei, Y. Lan, J. Chang, Nat. Commun. 2024, 15, 2833. doi: 10.1038/s41467-024-47169-9.
正文:
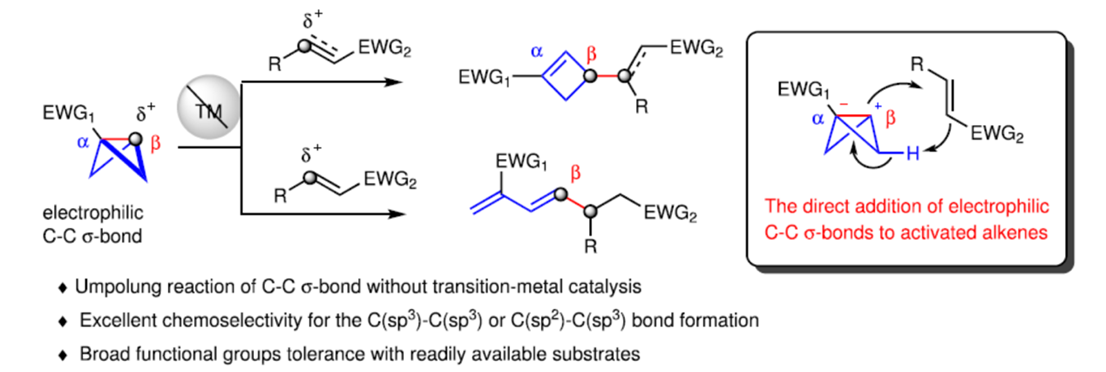
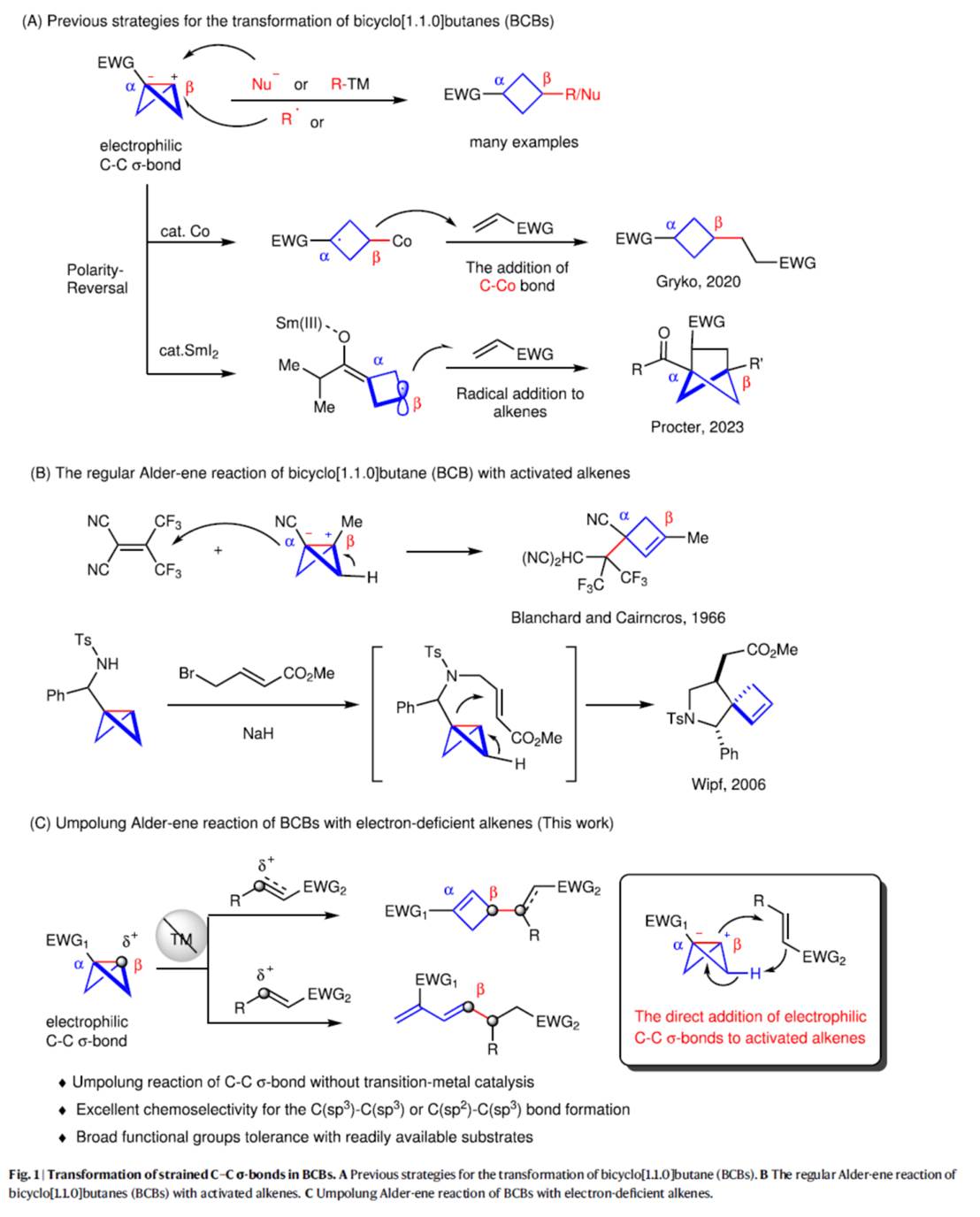
前期,诸多研究团队已经成功设计出多种利用bicyclo[1.1.0]butane (BCBs)构建官能团化环丁烷分子的合成转化策略[1]。然而,BCBs参与的极性反转反应方法学,目前却较少有相关的研究报道(Fig. 1A) [2]。受到BCBs中极化C-C σ-键与活化烯烃的加成反应方法学(Fig. 1B) [3]相关研究报道的启发,这里,河南师范大学的常俊标与白大昌等人报道一种全新的无金属催化bicyclo[1.1.0]butanes (BCBs)与缺电子烯烃的极性反转Alder-ene反应方法学,进而成功完成一系列官能团化的环丁烯与共轭二烯分子的构建 (Fig. 1C)。
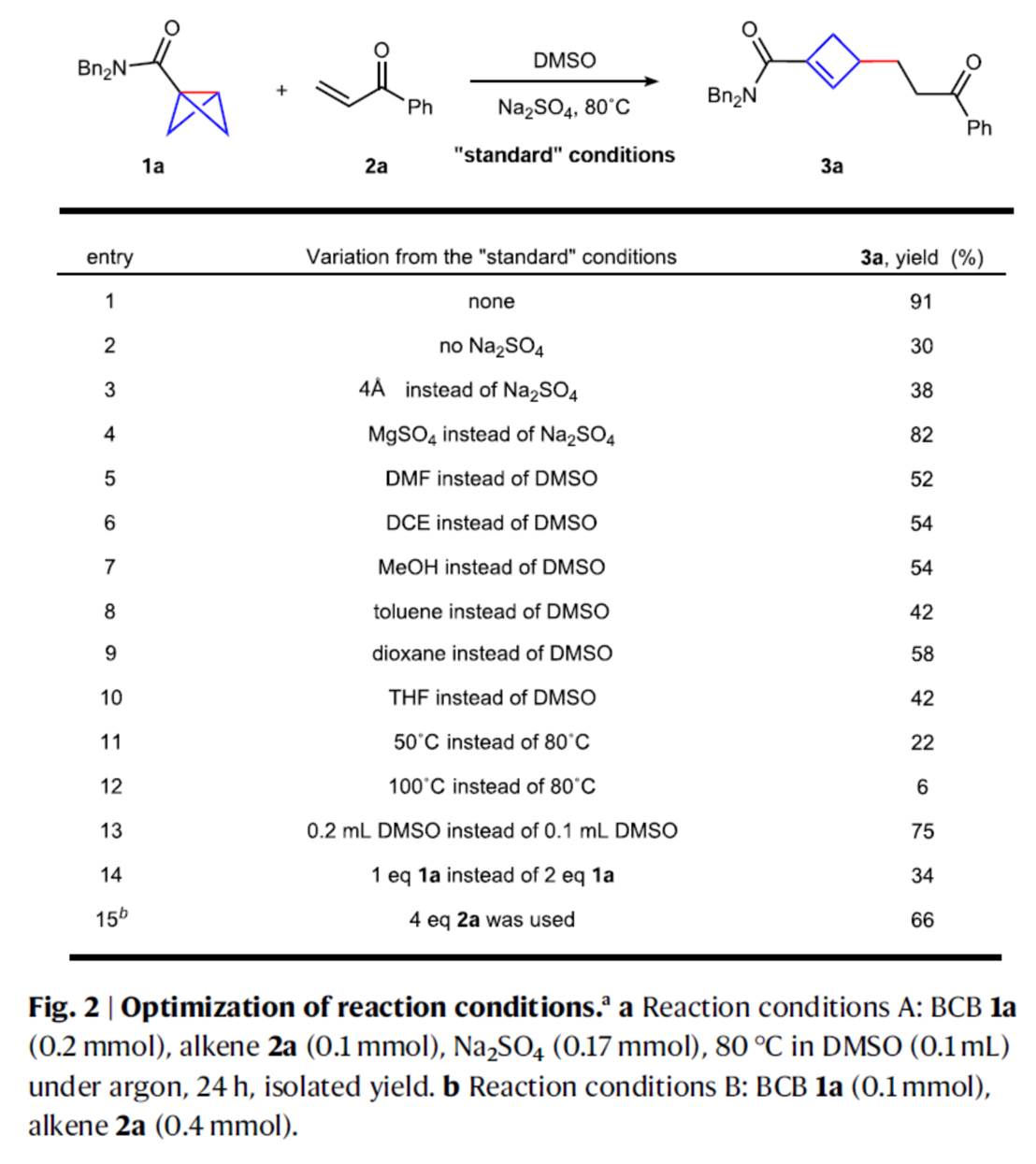
首先,作者采用bicyclo[1.1.0]butane (BCB) 1a与烯烃衍生物2a作为模型底物,进行相关反应条件的优化筛选 (Fig. 2)。进而确定最佳的反应条件为:采用Na2SO4作为添加剂,在DMSO反应溶剂中,反应温度为80 oC,最终获得91%收率的产物3a。
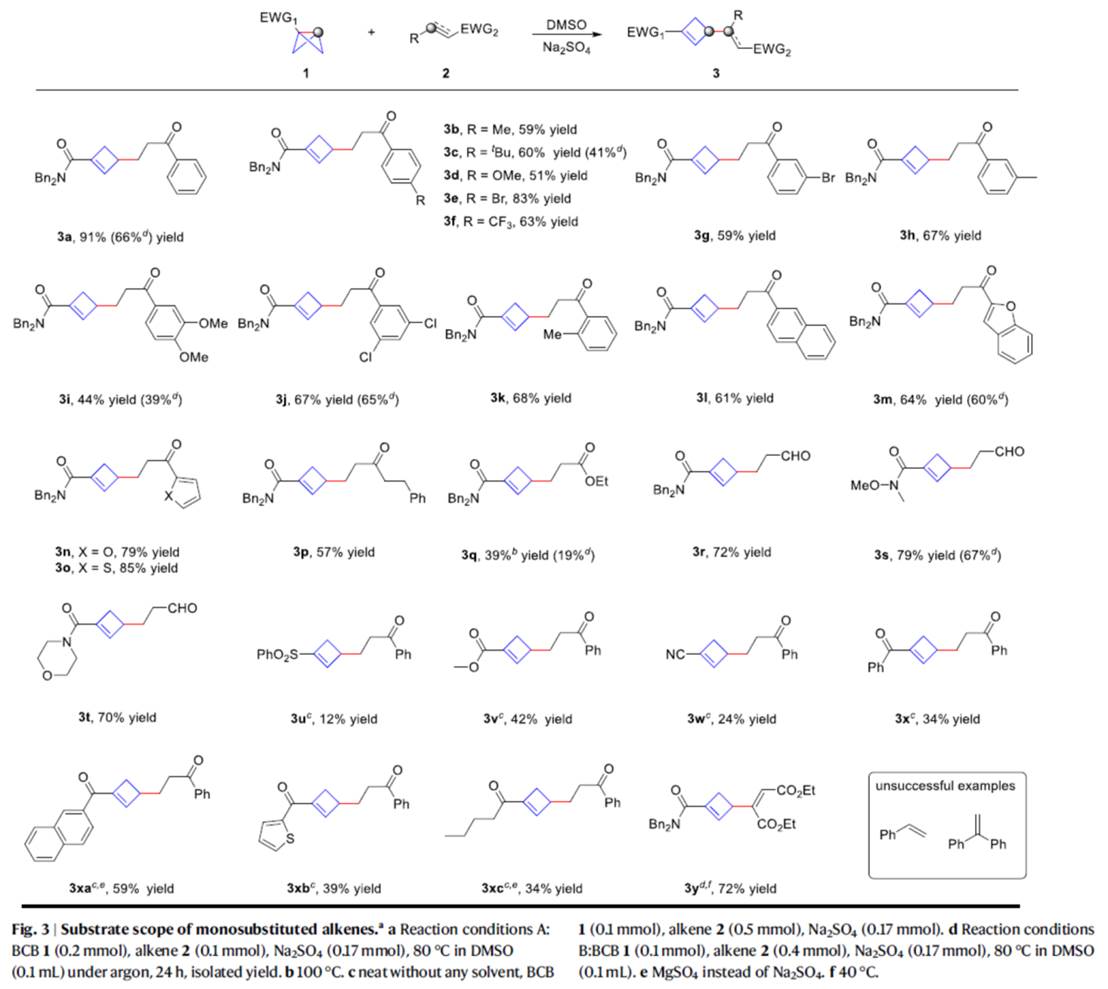
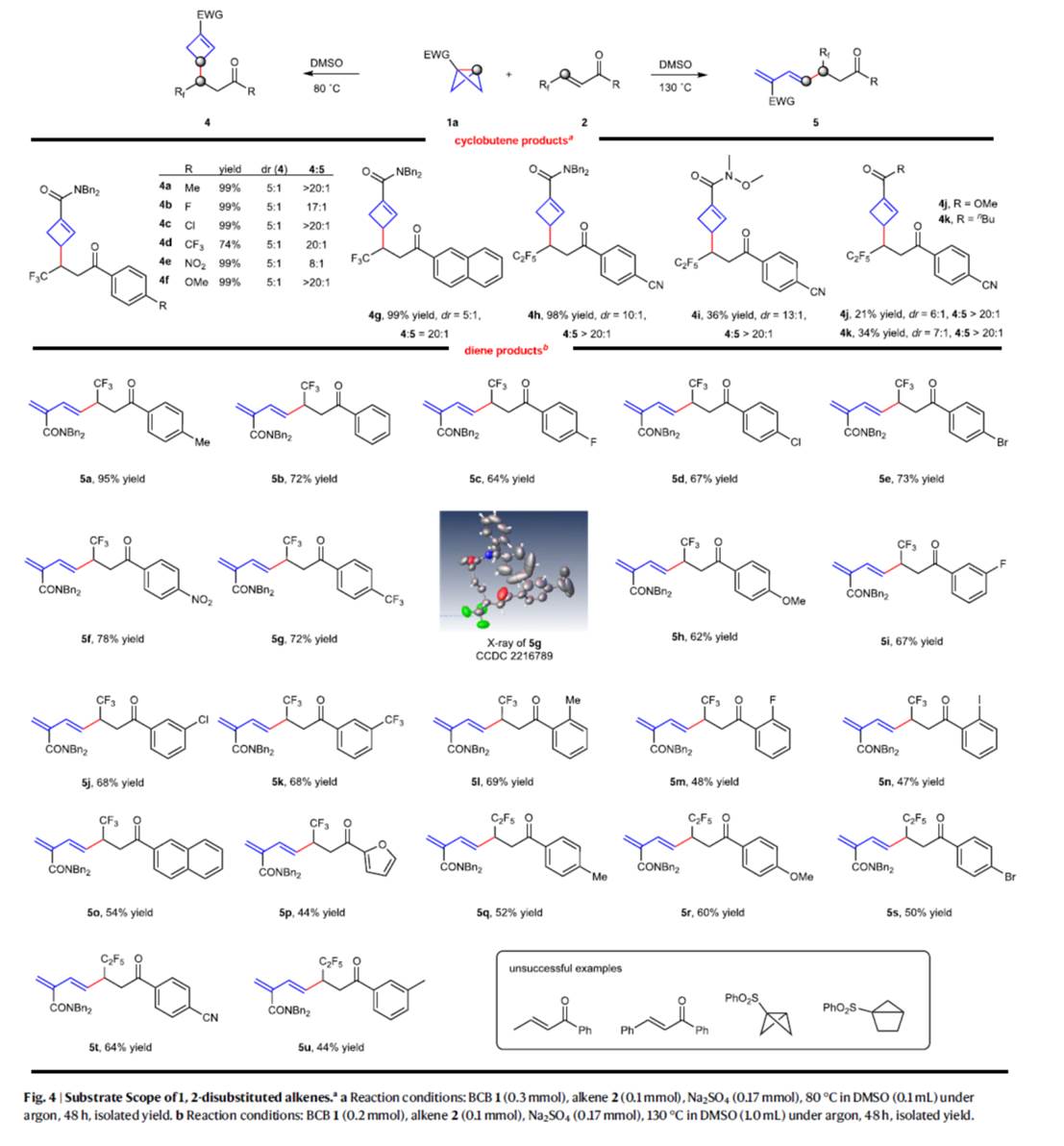
在上述的最佳反应条件下,作者分别对一系列单取代BCBs底物 (Fig. 3)以及1,2-二取代烯烃底物 (Fig. 4)的应用范围进行深入研究。


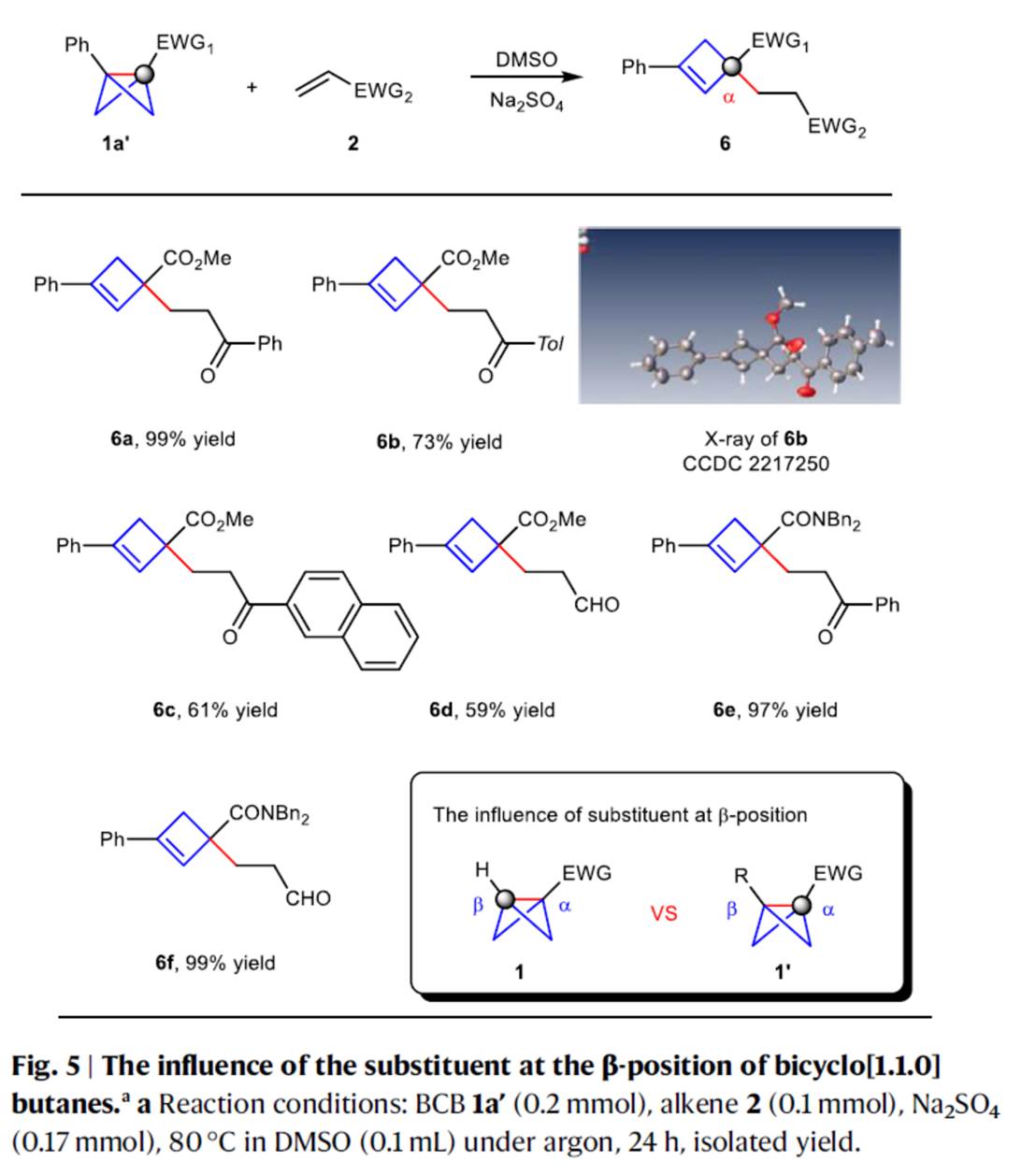
同时,作者还对BCBs中β-位的取代基对极性反转反应的影响进行了研究(Fig. 5)。

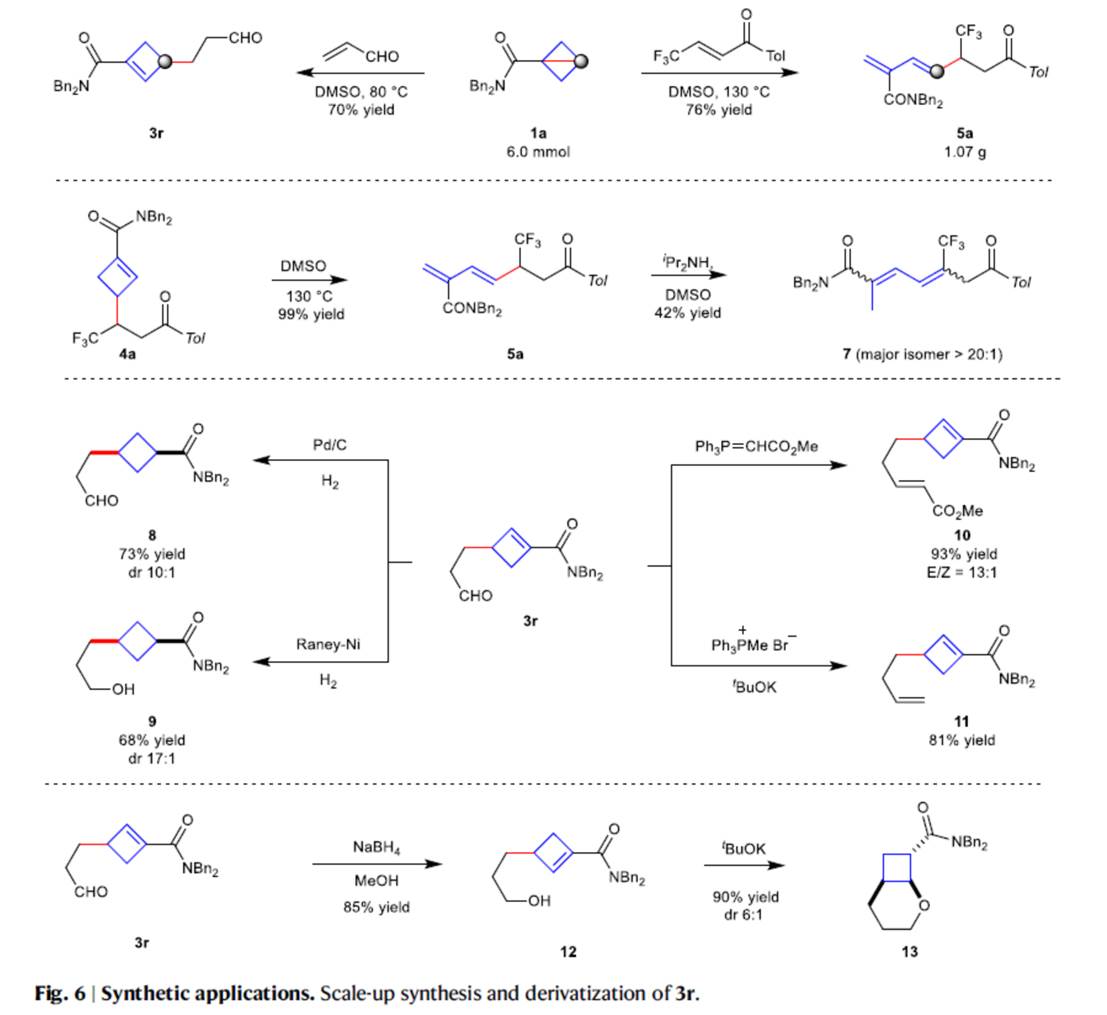
之后,该小组通过如下的一系列研究进一步表明,这一全新的极性反转Alder-ene策略具有潜在的合成应用价值 (Fig. 6)。
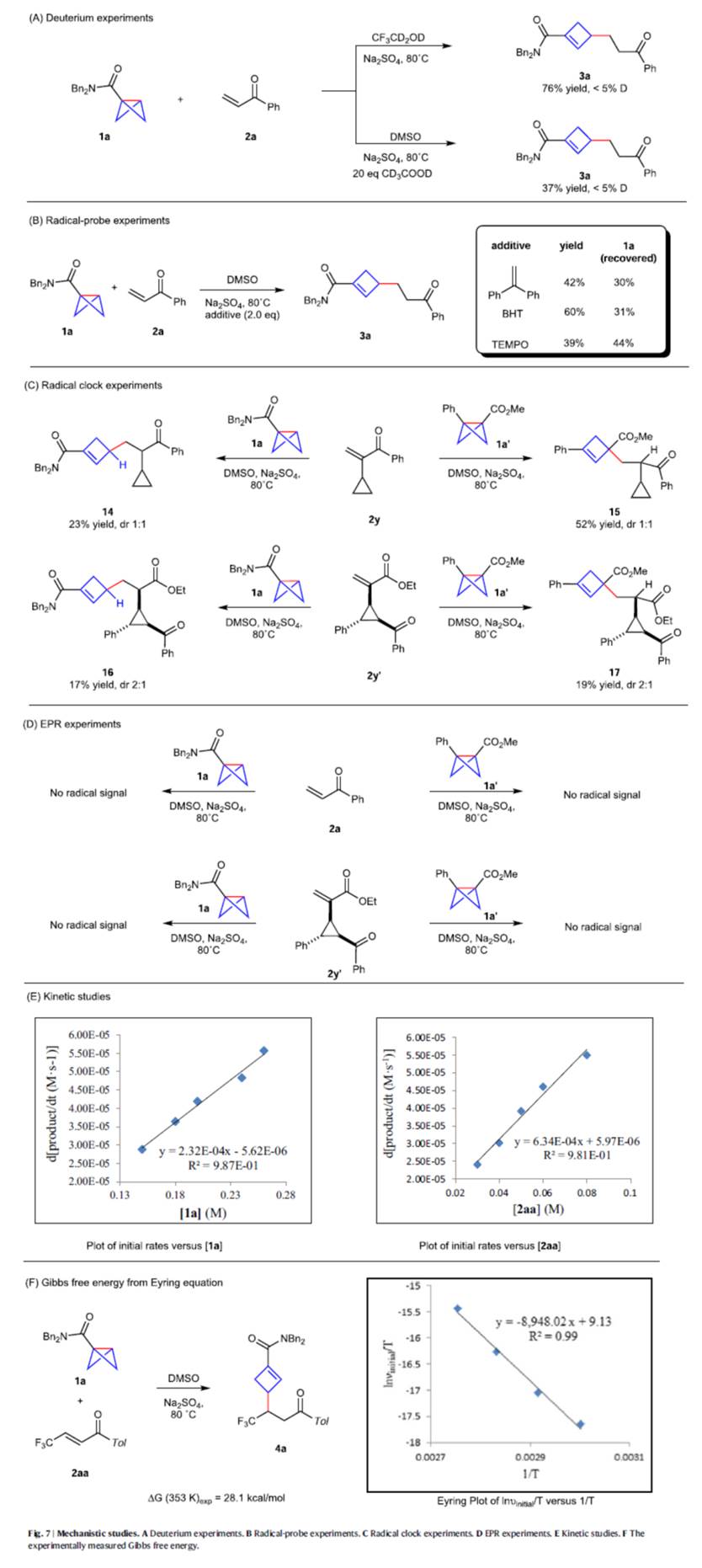
接下来,作者对上述极性反转Alder-ene过程的反应机理进行进一步研究 (Fig. 7)。

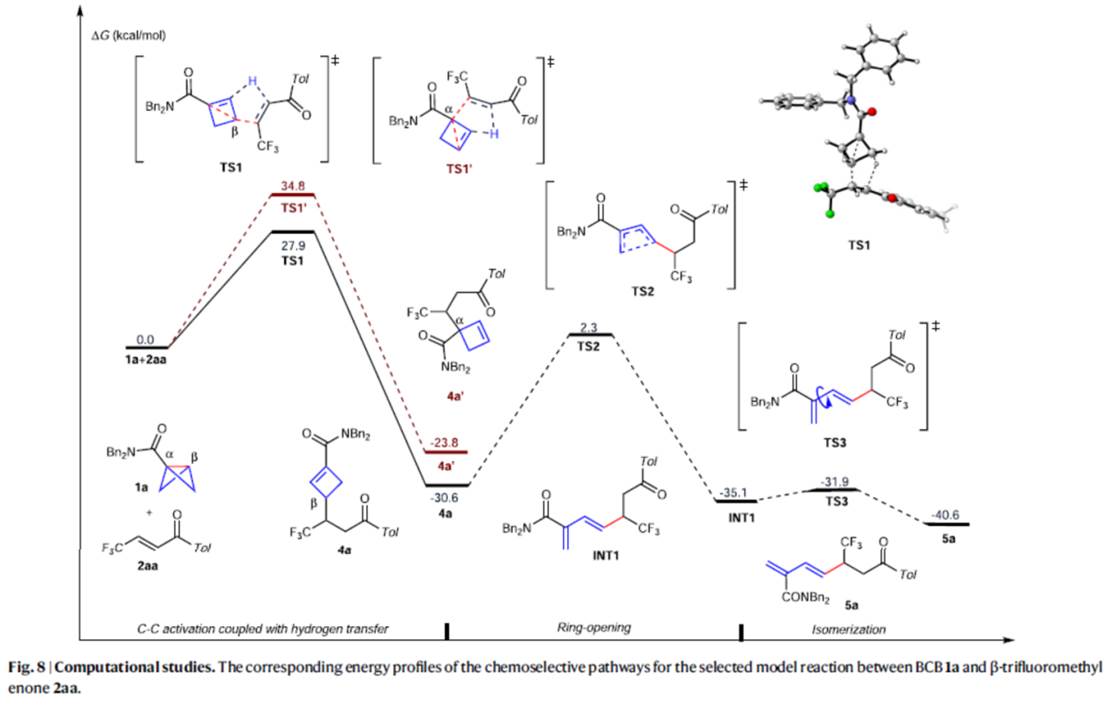
此外,作者还对反应的过程进行了相关的理论计算研究 (Fig. 8)。
总结:河南师范大学的常俊标与白大昌等人报道一种全新的无金属催化bicyclo[1.1.0]butanes (BCBs)与缺电子烯烃的极性反转Alder-ene反应方法学,进而成功完成一系列官能团化的环丁烯与共轭二烯分子的构建。这一全新的极性反转合成转化策略具有底物范围广泛、优良的官能团兼容性、优良的原子经济性以及优异的选择性等优势。
参考文献:
- [1] A. Fawcett, A. Murtaza, C. H. U. Gregson, V. K. Aggarwal, J. Am. Chem. Soc. 2019, 141, 4573. doi:10.1021/jacs.9b01513.
- [2] M. Ociepa, A. J. Wierzba, J. Turkowska, D. Gryko, J. Am. Chem. Soc. 2020, 142,. 5355. doi:10.1021/jacs.0c00245.
- [3] P. G. Gassman, Acc. Chem. Res. 1971, 4, 128. doi:10.1021/ar50040a002.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
















No comments yet.