
[导读]
过渡金属催化含氮杂环衍生物的不对称自由基官能化反应至今仍是一大难题。现有催化体系均无法实现强配位含氮杂环衍生物的不对称叠氮化,其根源在于这类强配位底物易使催化剂中毒。铜催化不对称自由基叠氮化领域中的另一难题是N3−与铜容易形成双叠氮Cu(II)物种,该物种是导致立体选择性差的重要因素之一。
近日,武汉大学陈才友/戚孝天团队在这一方向取得突破性进展——通过开发新型强配位手性三齿PNN配体,实现了强配位底物含氮杂环的不对称自由基叠氮化反应,有效抑制N-杂芳烃竞争配位导致的催化剂毒化,同时避免形成选择性的双叠氮Cu(II)物种,从根源上重塑了铜-叠氮体系的配位化学行为。该成果近期发表在Nature Catalysis上,武汉大学陈才友教授为论文通讯作者,戚孝天教授为论文共同通讯作者,武汉大学博士生雷一凡为第一作者。
Unlocking the Cu-catalysed asymmetric radical azidation of N-heterobenzylic sites through rational ligand design
Lei, Y.; Chen, Z.; Guo, H.; Yuan, Q.; Zhu, A.; Zhang, Z.; Bai, Y.; Qi, X.*; Chen, C*.
Nat. Catal. 2026. doi: 10.1038/s41929-026-01563-2
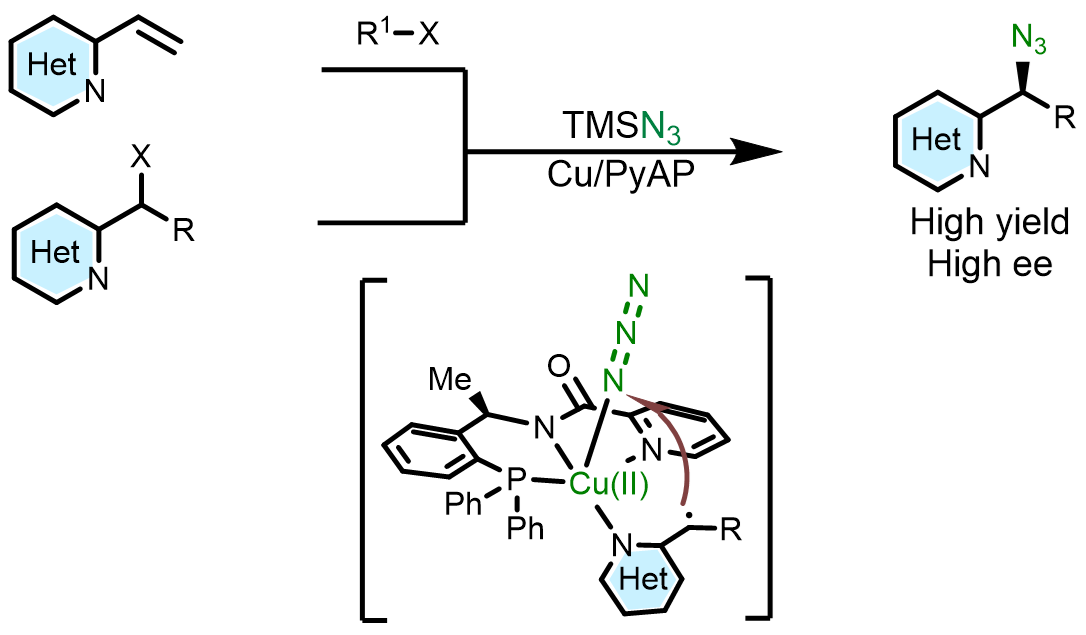
正文:
过渡金属催化含氮杂环衍生物的不对称自由基官能化反应至今仍是一大难题。现有催化体系均无法实现强配位含氮杂环衍生物的不对称叠氮化,其根源在于这类强配位底物易使催化剂中毒。铜催化不对称自由基叠氮化领域中的另一难题是N3−与铜容易形成双叠氮Cu(II)物种,该物种是导致立体选择性差的重要因素之一。
近日,武汉大学陈才友/戚孝天团队在这一方向取得突破性进展——通过开发新型强配位手性三齿PNN配体,实现了强配位底物含氮杂环的不对称自由基叠氮化反应,有效抑制N-杂芳烃竞争配位导致的催化剂毒化,同时避免形成选择性的双叠氮Cu(II)物种,从根源上重塑了铜-叠氮体系的配位化学行为。
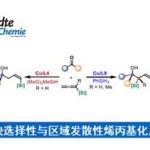
含氮杂环是广泛存在于各类天然产物、农用化学品、生物活性分子与药物分子中的通用骨架。近十年经美国食品药品监督管理局批准上市的小分子药物中,超过82%的分子均含有至少一个氮杂环。吡啶、嘧啶等含氮杂环可显著提升药物与靶点结合能力:将分子苯环骨架替换为吡啶骨架时,可使药效提升2倍,替换为2 -嘧啶基药效提升4倍(图1a)。叠氮根配位能力强,双齿配体配位下金属中心易结合两分子N3−,进而形成双叠氮 Cu (II) 中间体,烷基自由基可与任意叠氮基团发生反应,过渡态无手性区分,产物ee值极低(图1b)。为解决此问题,该团队设计和合成出新型强配位手性三齿PNN配体,该配体具有以下设计思路:一、三个配位点均为强配位点,可避免双叠氮 Cu (II)的形成;二、可被去质子化的酰胺配位点,在碱性条件下能与铜形成富电性的σ配位,增强配体与铜的结合能力;三、σ-配位作用拉近了配体的手性位点与铜原子的距离,易于对映选择性的控制。四、吡啶骨架的可修饰性能进一步提高对映选择性(图1d)。含有弱配位(hemi-labile效应)的NMe2取代的配体在反应体系中,该位点容易被体系中N3−取代而形成双叠氮二价铜物种,进而导致立体选择性变差(图1c)。含有强配位吡啶取代的配体在反应体系中,由于强配位特性,该位点不易被N3−取代,因而更易形成单叠氮二价铜物种,更有利于立体选择性的调控(图1e)。本研究利用强配位三齿配体,以优异的收率和对映选择性实现了铜催化2-(1-卤代烷)-含氮杂环的对映汇聚式不对称自由基叠氮化反应,以及铜催化2-乙烯基-含氮杂环的三组分不对称自由基叠氮化反应(图1f)。
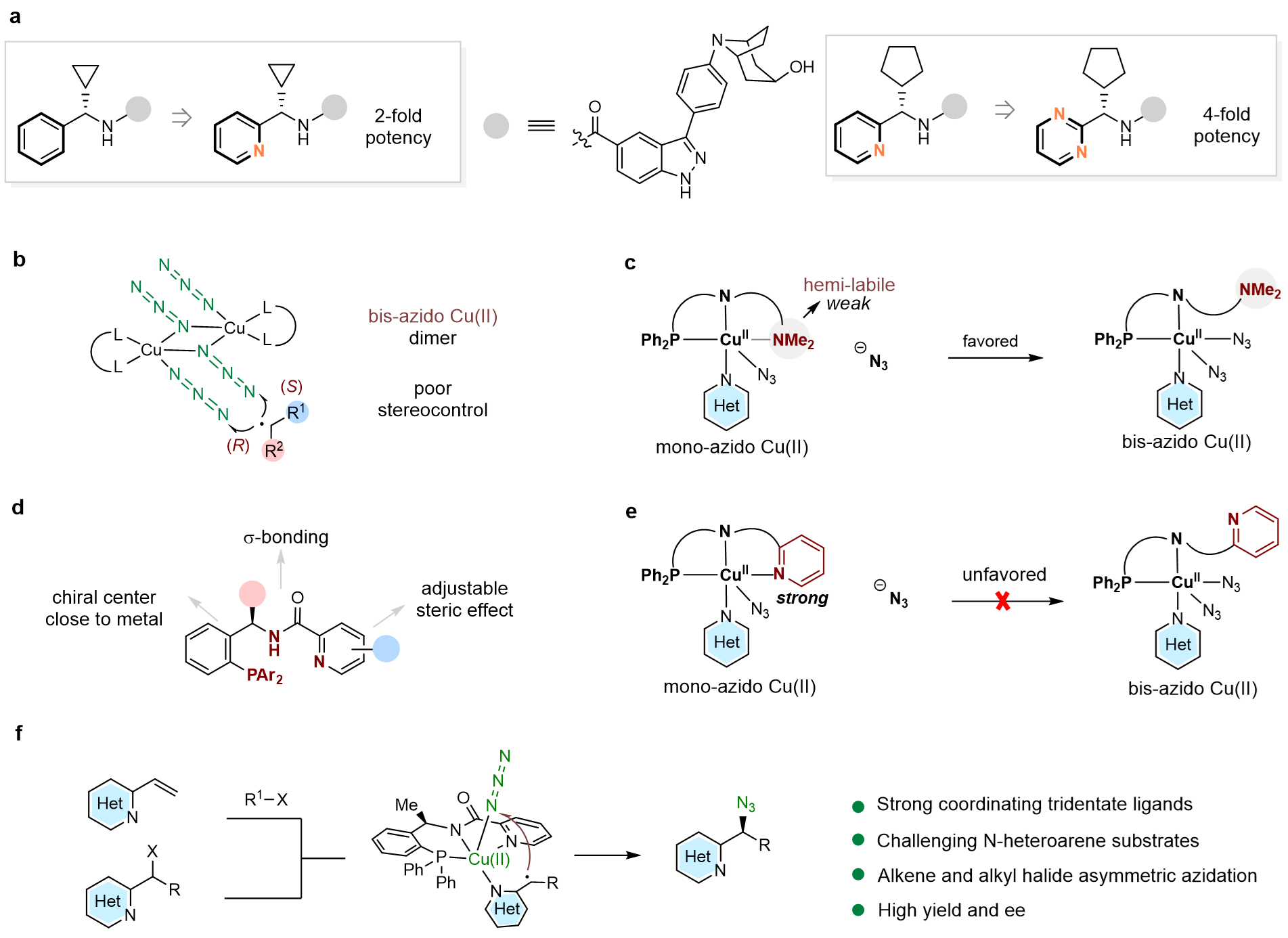
图1. 研究背景及本文策略
反应条件筛选中发现(图2),手性位点的空间位阻对反应立体选择性调控具有一定影响,结果显示,-Me为最佳取代基,当使用-Et(L1)、-Bn(L2)、-Ph(L3)取代的配体均降低ee值。-PR2上可修饰的取代基的空间位阻对反应对映选择性影响最大,结果显示,-PPh2为最佳取代基,其他大位阻配体(L4、L5、L6)均降低ee值。将配体吡啶氮的邻位引入取代基的配体(L7、L8)均不能促使反应的发生,原因是该位点的修饰会阻碍配体吡啶与Cu的配位,进而弱化了配体的配位性。将强配位吡啶改变为弱配位NMe2(L9)时,该配位点容易被催化体系中的强配位底物(含氮杂环)取代,而发生中毒失活现象。喹啉取代的配体(L10)由于位阻原因影响配体与金属配位而无法促使反应的发生。其他配体(L11、L12、L13)均无法促使强配位底物(含氮杂环)的不对称自由基叠氮化反应,原因可能是催化剂中毒导致。
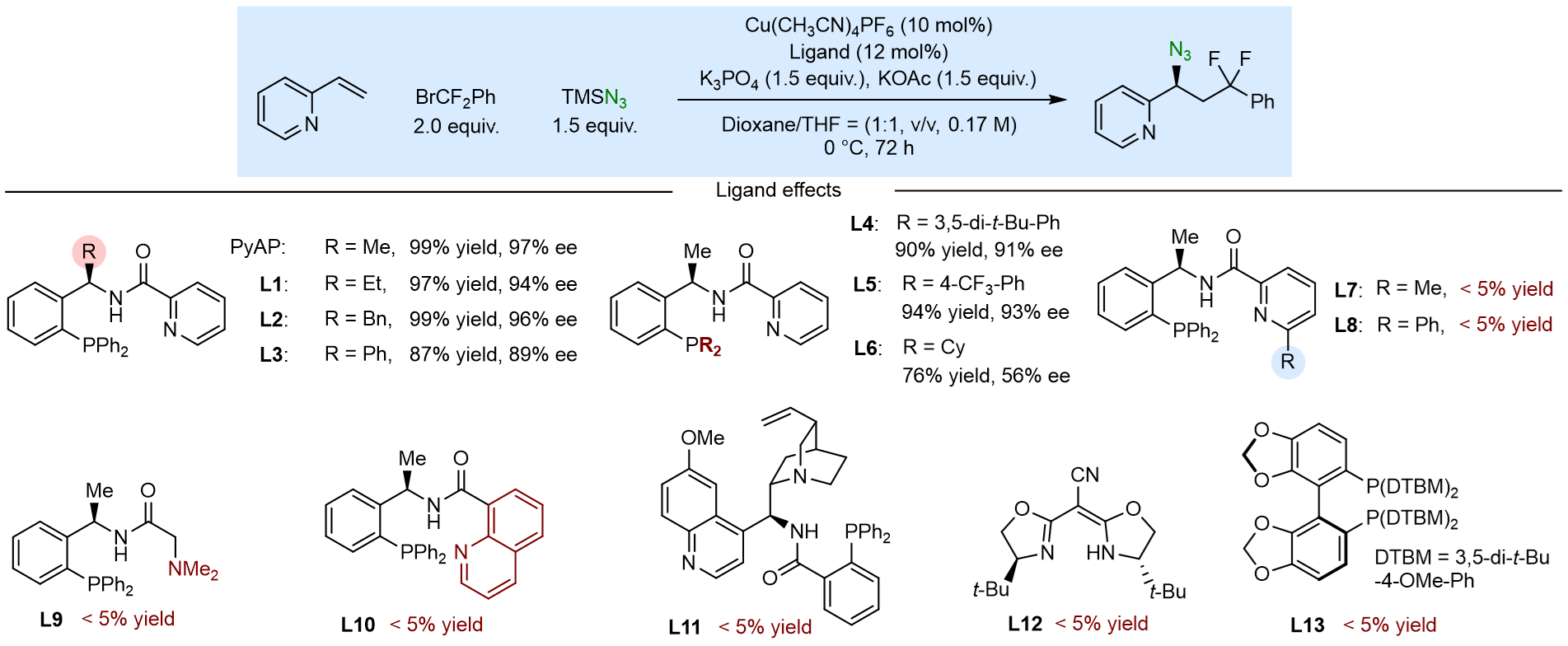
图2. 条件筛选
作者首先以BrCF2Ph为自由基前体,在最优条件下,考察了铜催化2-乙烯基-含氮杂环的三组分不对称自由基叠氮化反应的底物拓展情况(图3)。不加铜源、配体、碱均不能促使反应的发生;不加添加剂收率降低;催化剂负载量减半,收率降低但ee值基本不变。对于吡啶中C-4位和C-5位上引入吸电子取代基和供电子取代基的底物,均能以中等偏上的收率和优异对映选择性获得叠氮产物。若在吡啶上C-3位引入F取代基,收率和ee值均大幅度降低,原因是该取代位置的空间位阻会直接影响自由基和铜的相互作用。此外,该催化体系兼容Memantine、Metronidazole、Isoxepac、Naproxen、Diclofenac、Pleuromulin、Sulbactam等多种复杂分子,凸显了其在合成复杂功能分子方面的实用价值。更重要的是,该催化体系兼容包括吡啶在内的11种强配位含氮杂环,且这些底物均未毒化催化剂。此外,作者考察了15种自由基前体,包括11种二氟溴代烷、1种二氯氯代烷、3种非二氟类溴代烷。
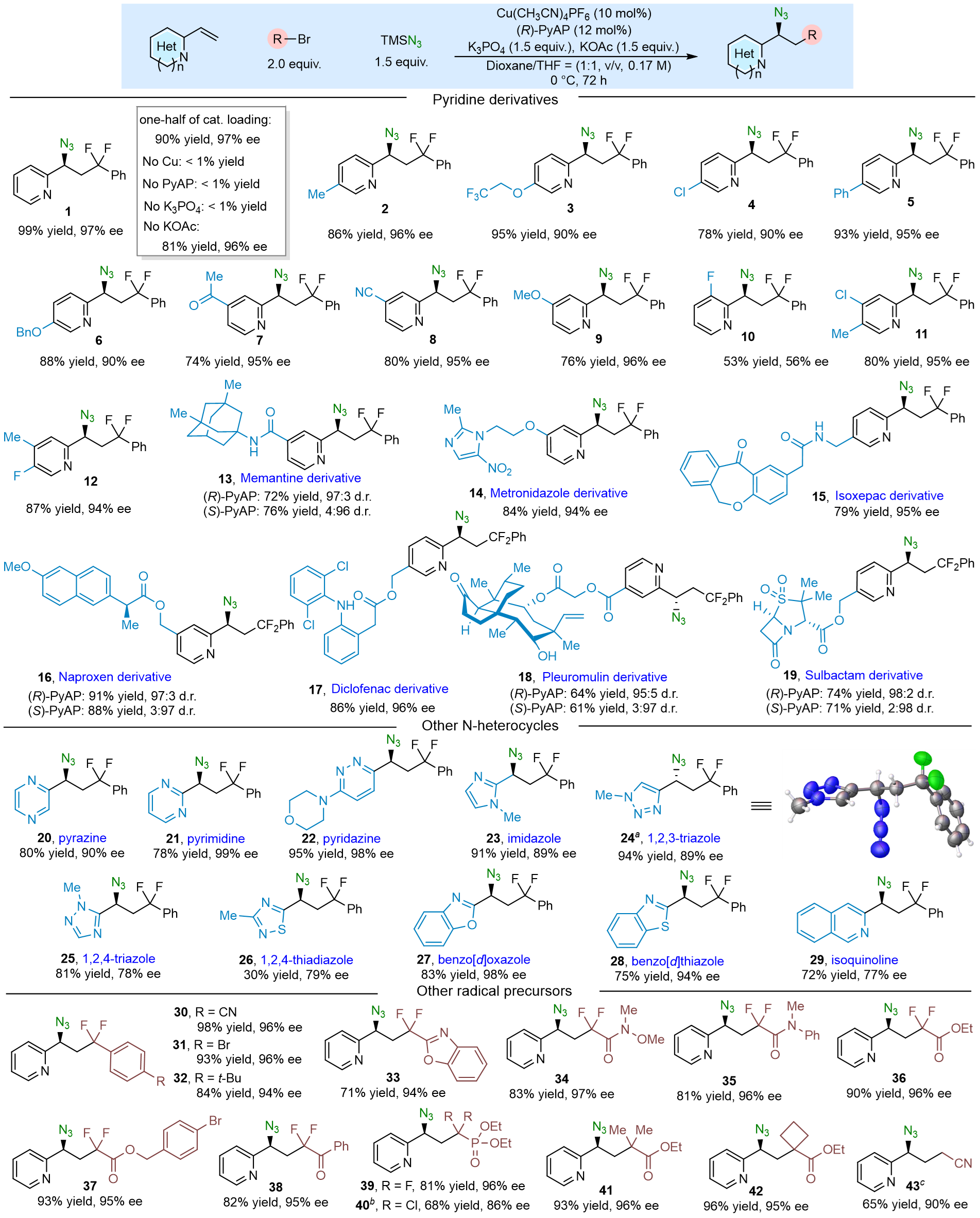
图3. 2-乙烯基-含氮杂环的底物拓展
之后,作者考察了铜催化2-(1-卤代烷)-含氮杂环的对映汇聚式不对称自由基叠氮化反应的底物拓展情况(图4)。该反应不仅兼容烷基溴代物,而且兼容烷基氯代物和烷基碘代物。对于直链的烷基侧链和环状的烷基侧链的底物,该催化体系同样兼容。大位阻取代基底物(环丁基、异丙基、叔丁基等)均能获得优异的对映选择性。同时,羰基β-溴代物,在该催化体系下并未生成α,β-不饱和羰基化合物。在不同取代基的吡啶杂环底物拓展方面,吡啶C-4位、C-5位不同修饰(包括吸电子基和供电子基)的底物均可在催化体系中获得优良的收率和对映选择性。Dettol、L-Menthol、D-glucose等复杂分子亦能适用于该催化体系。在含氮杂环类型方面,该方法兼容噻唑、嘧啶、吡嗪、苯并噁唑等含氮杂环,且这些底物均未毒化催化剂。
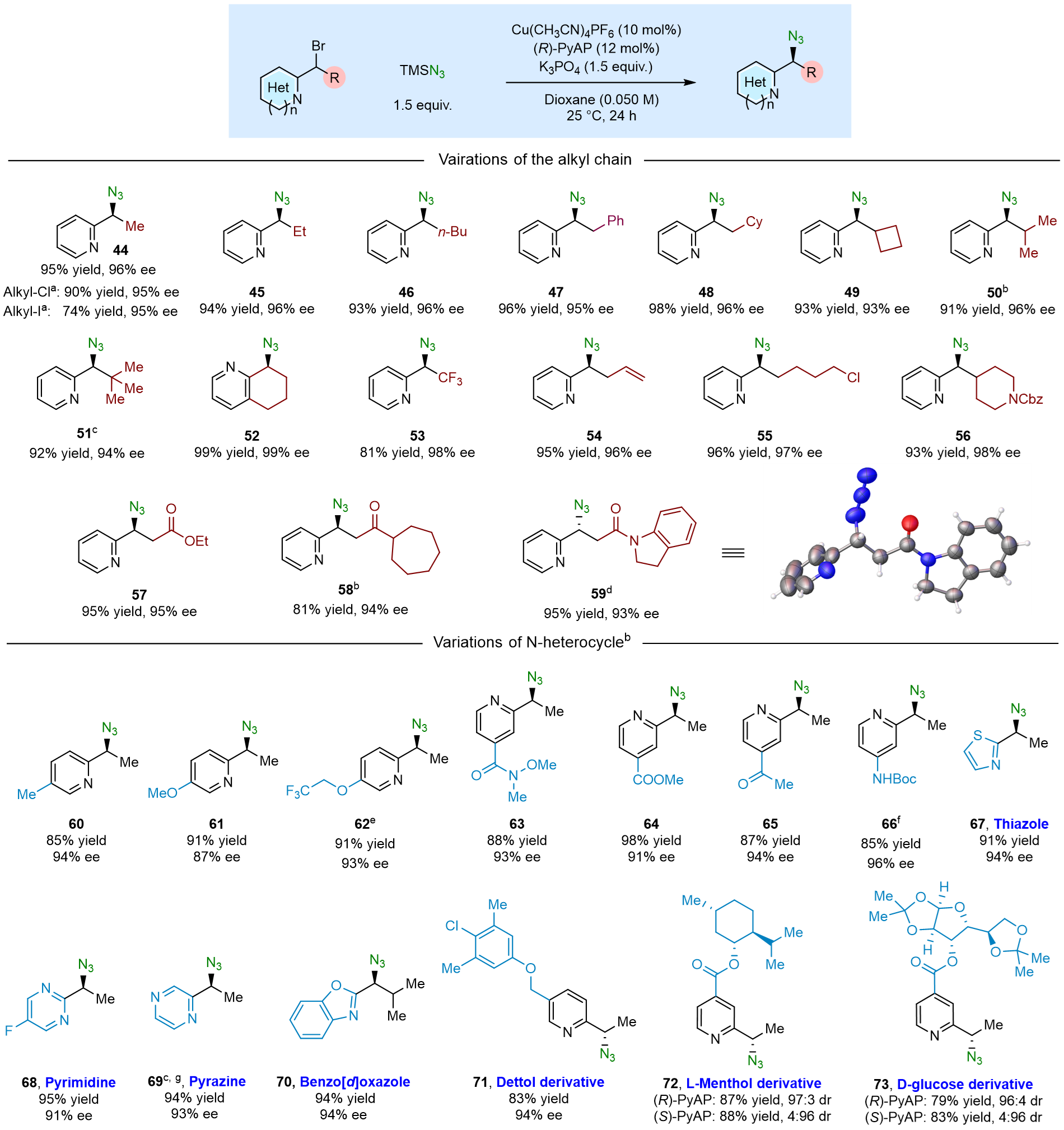
图4. 2-(1-卤代烷)-含氮杂环的底物拓展
转化与应用方面(图5),作者首先实现了催化反应的克级规模的放大,并且对映选择性不受影响,体现出该催化剂的优越性和立体控制性。其次,作者对手性叠氮官能团进行了应用转化,包括生成手性磷酰胺、手性三氮唑、手性酰胺等。值得一提的是,原料在PPh3/CS2条件下生成吡啶去芳构化后的咪唑−硫醇产物。作者利用生物活性分子5-乙炔基-2′-脱氧胞苷(可用于DNA标记)与手性叠氮进行Click反应生成可能具有同样潜在的生物活性的手性三氮唑产物。同时,该方法适应于5个生物活性分子或药物中间体的合成(非炎性蛋白-1抑制剂(Vanin-1 inhibitor)、昂唑司特(Ontazolast)、天然产物(Conulothiazole A&B)、钙离子通道拮抗剂(TTA-A8)、组蛋白去乙酰化酶抑制剂(NK-HDAC-1)、Jak/Start通路抑制剂(AZD1480)等),进一步表明了该反应具有极大的应用价值。
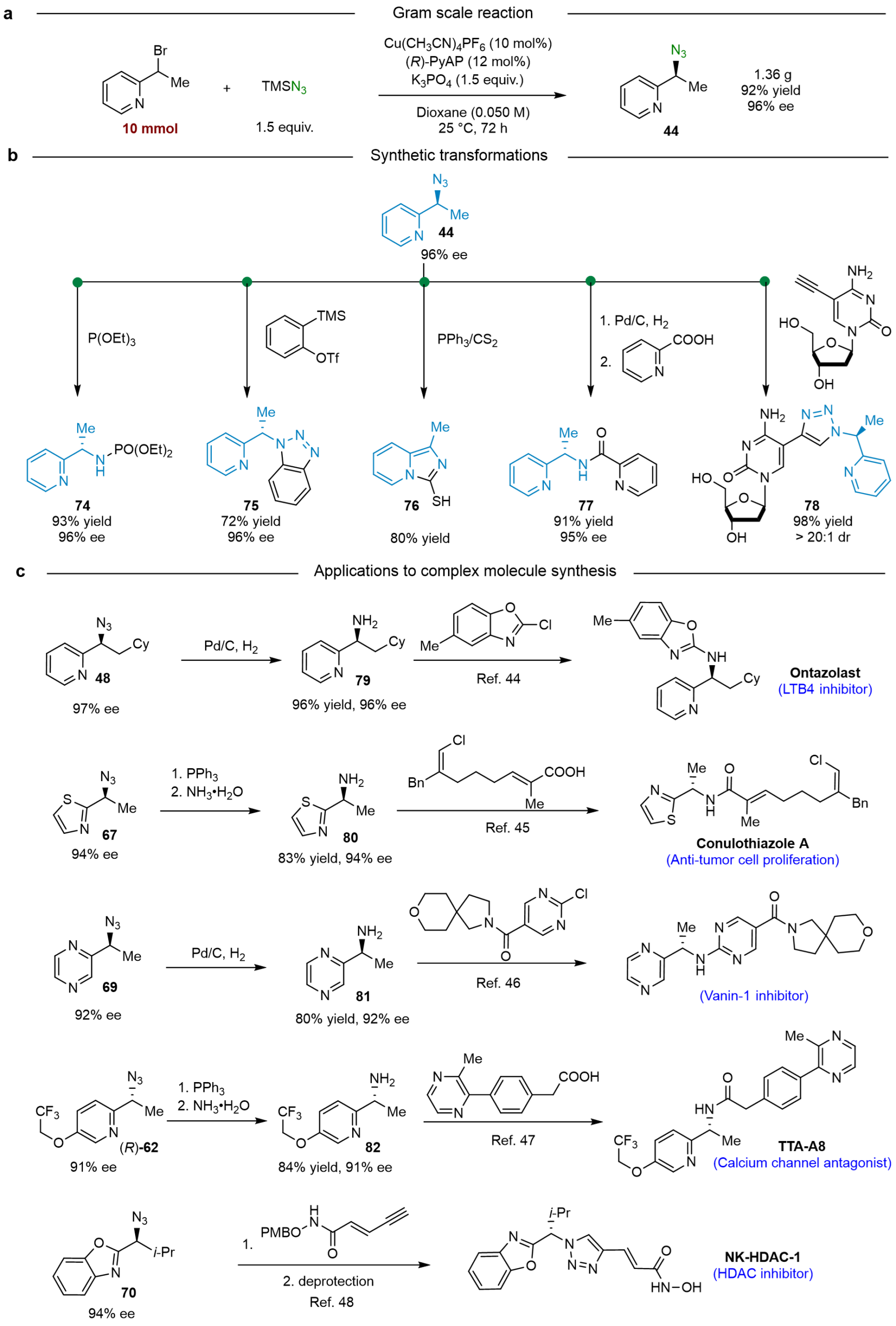
图5. 转化与应用
作者通过系列实验与理论计算,深入阐释了反应机理。
- TEMPO捕获实验成功分离到吡啶苄基自由基加合物(8% 收率);监测催化反应2 h的数据表明铜催化2-(1-卤代烷)-含氮杂环的不对称自由基叠氮化反应是对映汇聚式过程(图6a);该催化反应排除了自由基—极性交叉反应路径(图6b);在n-Bu3SnH/BEt3的常规自由基体系中未检测到环化产物,且生成51%的链状产物。而在铜参与的催化反应中,2.0 mol%的催化剂即可生成10%的环化产物,两者结果比较说明环化产物的生成必须有铜催化剂的参与。其次,随着铜催化剂量的增加,U/C线性降低,进一步表明烷基自由基可能参与了铜的配位,进而生成环化产物(图6c)。
- 动力学数据表明该反应对催化剂为一级反应,对亲电试剂为零级反应,对亲核试剂(TMSN3)为一级反应,对K3PO4当量为一级反应。作者平行开展了三组KIE实验,平均值kH/kD值为0.81,含氮杂环苄位碳自由基与N3−成键反应(内球层均裂取代(Inner-sphere SH))为决速步,其中自由基上的碳原子从sp2转变为sp3(图6d)。
- 作者将非强配位的吡啶底物更换为苯环时,反应仅能生成消旋体,表明底物氮原子需与铜配位,才能发生不对称自由基叠氮化反应。非线性效应实验表明铜与配体在催化反应中以1:1(摩尔比)形成催化剂,并参与催化循环。Time-course实验表明催化过程中没有动力学拆分过程,且支持了对映汇聚式过程。
- EPR实验表明该催化体系中存在5配位二价铜模型(图7a),该模型表明三齿配体、含氮杂环、N3−均配位在Cu(II)中心,通过与底物发生可逆互换(图7b,D与E),进而发生分子内成键。同时EPR实验进一步表明弱配位配体易形成双叠氮二价铜物种,强配位配体易形成单叠氮二价铜物种。并且核磁滴定和EPR实验共同解释了N3−是在Cu(II)阶段交换到铜上,而不是Cu(I)阶段。
戚孝天团队对该工作进行了系统的DFT计算,计算数据支持XAT路径,并排除了氧化加成途径;计算支持内球层均裂取代过程,同时排除了外球层均裂取代反应途径,原因是(S)-TS-3b能垒(S)-TS-3高1.9 kcal/mol。决速步是内球层均裂取代过程,而不是XAT过程,原因是(S)-TS-3能垒TS-1高5.3 kcal/mol。DFT数据支持对映选择性控制,原因是(R)-TS-3的能垒仅比(S)-TS-3高出1.7 kcal/mol。同时,计算数据发现,强配位PNN配体中的吡啶配位点被底物替换、自身解离、N3−的替换均是不利吸热过程,从而支持了“新型强配位PNN配体可防止催化剂中毒”这一创新点(图8)。
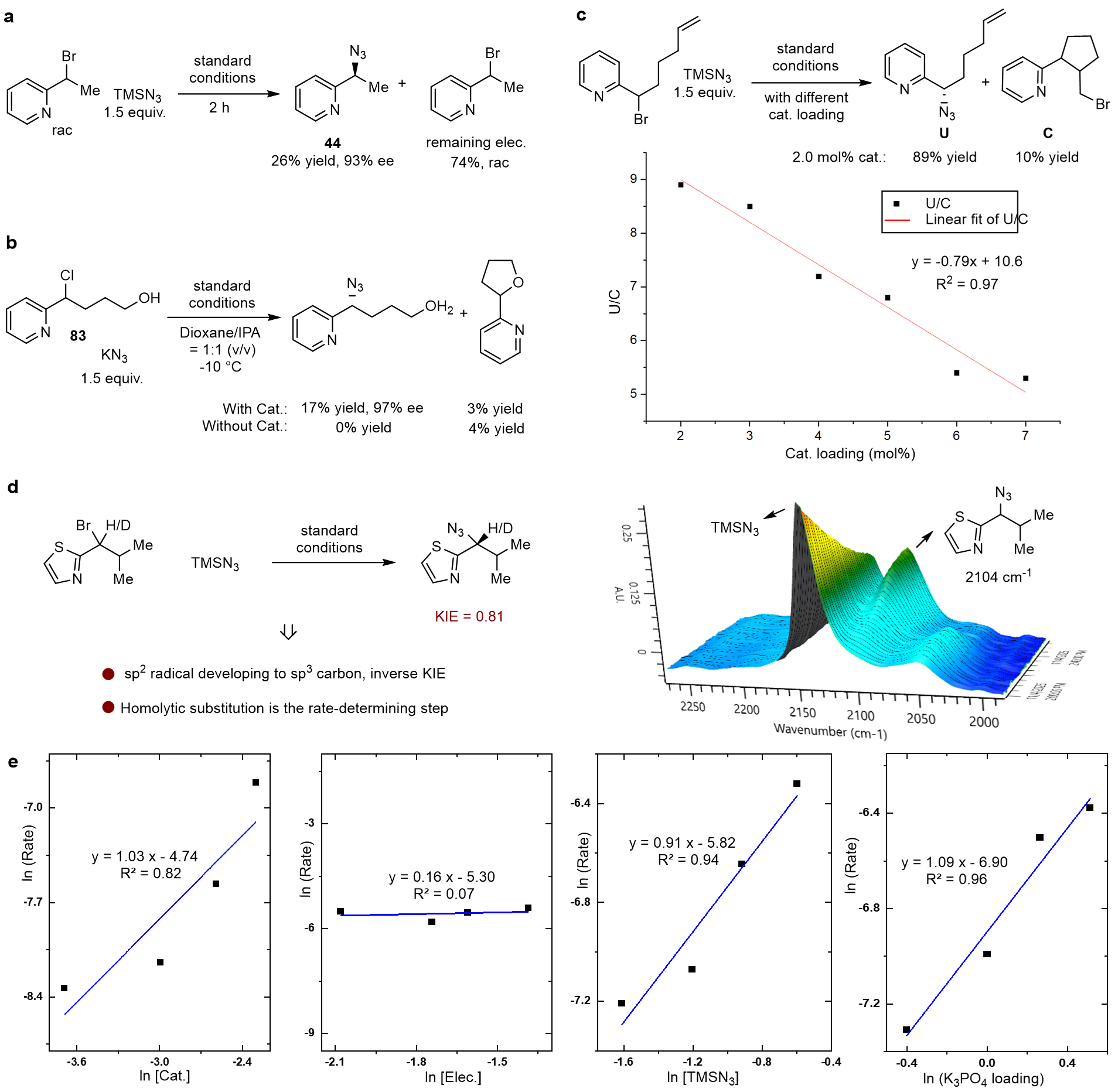
图6. 机理实验
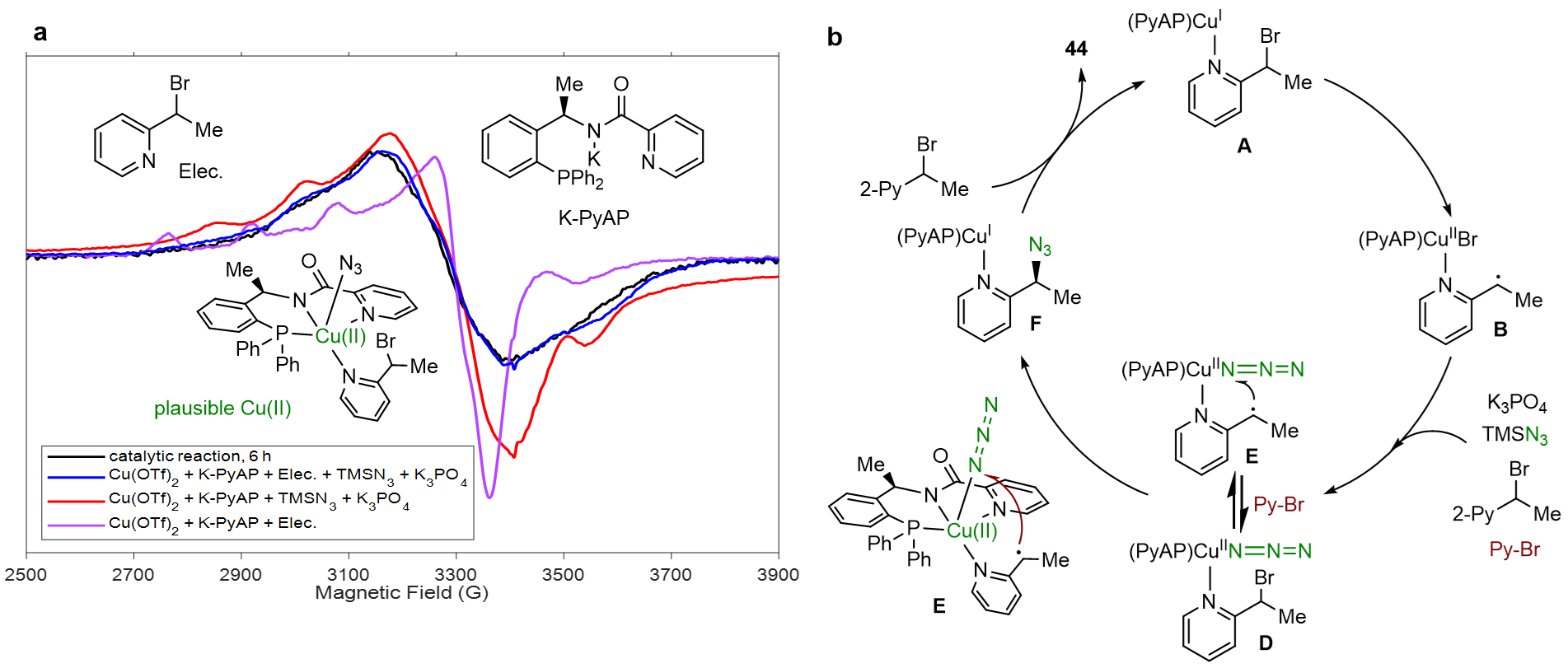
图7. EPR实验和推测的机理

图8. DFT计算
[导师介绍]
陈才友教授简介

陈才友,武汉大学化学与分子科学学院教授,博士生导师,课题组长,国家重点研发计划青年项目首席科学家, Chin. J. Chem.青年编委。2022年入选国家海外高层次人才引进计划,同年入选湖北省海外高层次人才项目,2023年入选武汉英才。2012年本科毕业于武汉大学,2017年博士毕业于武汉大学,师从张绪穆教授。2017~2022年,在美国加州理工学院从事博士后研究,师从美国科学院院士Gregory C. Fu和Jonas C. Peters。迄今在Nature (2篇); Nat. Catal. J. Am. Chem. Soc.; Angew. Chem. Int. Ed.; Acc. Chem. Res.; Chem. Sci.等杂志上发表论文30余篇,同时授权6项专利。2017年,受邀参加第67届德国Lindau诺贝尔奖获得者大会;2022年,受邀参加第四届“世界顶尖科学家大会”。曾荣获2017年度“武汉大学十大学术之星”称号。2025年荣获“新和成”-《中国化学》创新奖。
研究方向:不对称光/电催化;生物质平台分子转化;手性配体及光敏剂开发; DFT计算;机理研究。
陈才友课题组链接:https://caiyouchen.whu.edu.cn/index.htm
陈才友课题组长期招募优秀的博士后研究员、博士及硕士研究生和研究助理,享受武汉大学相关福利政策,待遇非常优厚。课题组每年有足够博士及硕士研究生招生指标与学校重点资助博士后指标,欢迎对不对称光/电催化、手性催化剂开发、生物质平台分子转化和DFT计算等有浓厚兴趣的有志青年加入我们的研究小组。
博士后待遇按照科研背景和研究计划进行评定,年薪36-40万,特别优秀的待遇可以进一步协商,享受武汉大学博士后福利待遇(博士后公寓、公费医疗、子女入学、住房公积金等)。可申请武汉大学“卓越博士后计划”(年薪40万)、“重点博后资助计划”(课题组有两个名额)以及国家层次的博士后创新人才支持计划。















No comments yet.