
作者:石油醚
导 读
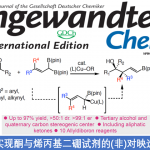
近日,受金属加氧酶次级配位环境中的氢键作用启发,济南大学王斌课题组报道了一种次级配位环境的氢键策略,在不需要强Lewis酸或Brønsted酸对碱性氮原子进行预先保护的情况下实现了非血红素锰配合物催化含氮分子中远端非活化C(sp3)‒H选择氧化。
“Hydrogen Bonding-Assisted and Nonheme Manganese-Catalyzed Remote Hydroxylation of C‒H Bonds in Nitrogen-Containing Molecules
Jie Chen, Wenxun Song, Jinping Yao, Zhimin Wu, Yong-Min Lee, Yong Wang,* Wonwoo Nam,* and Bin Wang*
J. Am. Chem. Soc.2023, ASAP. doi: 10.1021/jacs.2c13832.”
正 文
近年来,仿生催化非活化C(sp3)‒H选择氧化已成为改变天然产物和生物活性分子物理和生物学性质最直接有效的方式之一。依据化学键的强度、催化剂或底物的电子效应、立体效应及立体电子效应、导向基团、以及催化剂的绝对手性等固有因素,White、Costas、以及中科院兰州化物所孙伟研究员等报道了非血红素金属配合物催化甲基、亚甲基及次甲基C(sp3)‒H的位点及立体选择性氧化反应。除此之外,极性反转和分子识别也被用来调控C(sp3)‒H氧化的固有活性和选择性。经过近15年的发展,仿生催化C(sp3)‒H氧化取得了系列进展,但现有方法距离在合成上的广泛应用还存在诸多亟需解决的难题,例如,如何提高催化氧化的官能团兼容性,尤其是对于药物及农药研究至关重要的碱性氮官能团。
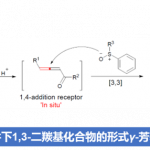
非血红素高价金属-氧中间体介导的含氮分子中脂肪族C‒H氧化的挑战性包括:(1)氮原子与金属中心的配位会导致催化剂失活(图1A),即碱性氮原子与金属中心的不可逆螯合会造成八面体金属中心的配位饱和,导致无法生成作为催化反应活性中间体的高价金属-氧物种;(2)化学选择性难以控制,即具有强亲电反应活性的高价金属-氧物种优先氧化碱性氮原子生成N-氧化物,而非脂肪族C‒H活化;(3)区域选择性难以控制,与远端C‒H相比,超共轭活化使得邻近N原子的α-C‒H官能化更容易发生。
以O2为氧源,自然界中的金属氧化酶在绝对温和、绿色的条件下催化脂肪族C‒H化学、区域和立体选择性氧化。在这一催化转化过程中,高价金属-氧中间体的生成以及对于活性位点物理和化学性质的精准调控是实现酶催化高效性和高选择性的关键。活性位点的物理和化学性质由主配位环境(以共价键的方式与金属中心结合的配体)和次级配位环境(活性位点内的非共价键相互作用)共同控制(图1C),前者决定几何构型、Lewis酸性、电子结构等基本性质,后者决定可及性和位点选择性。氢键是金属加氧酶用来调节催化金属中心周围环境最常见的非共价相互作用类型,研究证明次级配位环境的氢键不仅可以调节活性中心的静电性质,还能影响质子和电子的转移,控制金属介导的O2活化,引导底物的定位(图1C)。受金属氧化酶次级配位环境中的氢键作用启发,近日该课题组报道了一种溶剂氢键策略,以廉价易得的非血红素锰配合物作催化剂、以强氢键供体六氟异丙醇(HFIP)作溶剂、以H2O2作氧源,在不需要强Lewis酸或Brønsted酸对碱性氮原子进行预先保护(图1C)的情况下实现了含氮分子中远端非活化C(sp3)‒H选择氧化(图1D)。
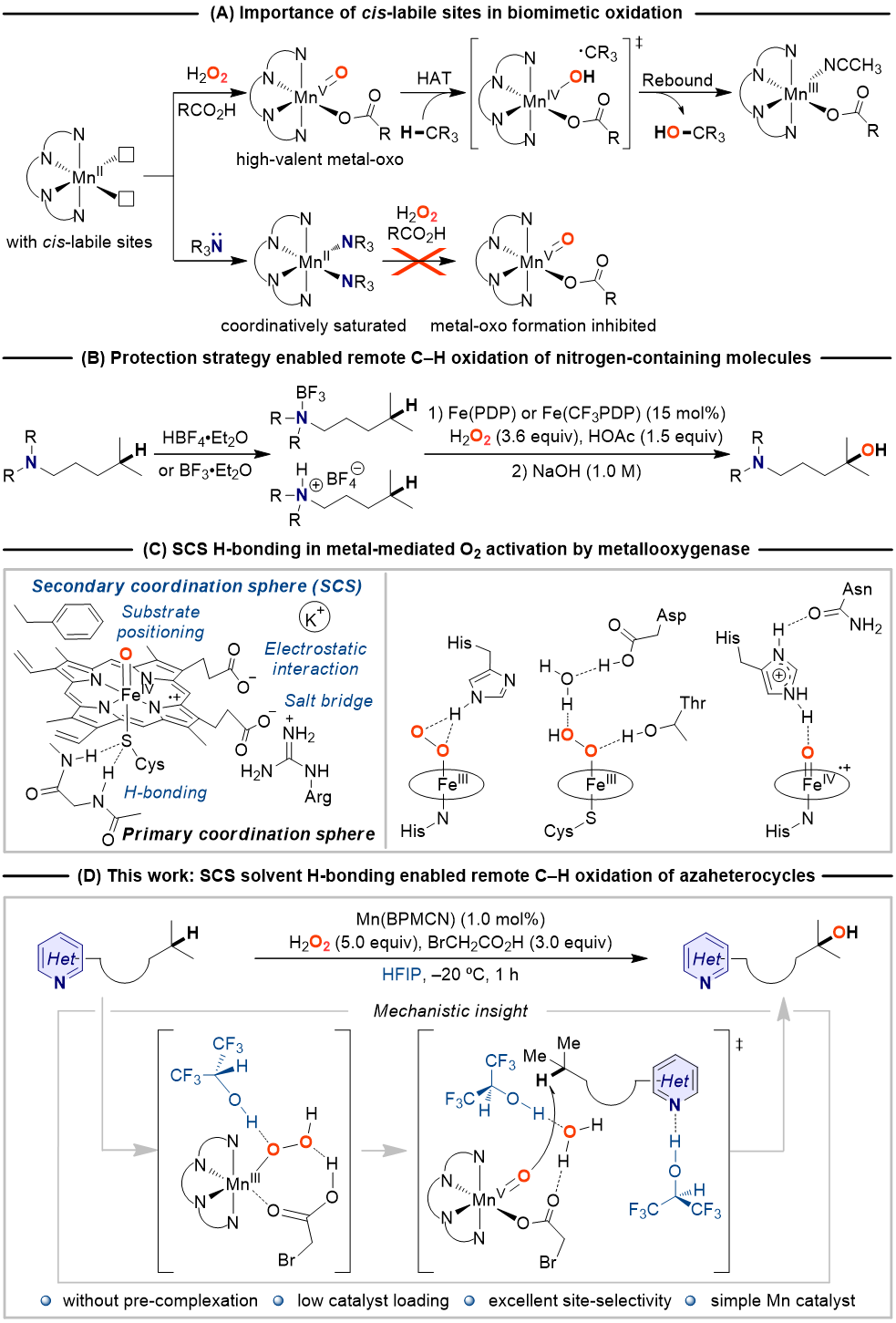
图1. 研究背景及工作设想
首先,作者系统研究了溶剂对Mn(BPMCN)催化2-4-甲基戊基吡啶1a选择氧化的影响(图2)。在以1.0 mol% Mn(BPMCN)作催化剂、5.0当量H2O2作氧化剂、3.0当量BrCH2CO2H作添加剂、六氟异丙醇(HFIP)作强氢键供体溶剂的条件下获得了最佳反应条件,在‒20°C下反应1小时,目标产物1b的产率为69%;未发现N-氧化物和α-C–H氧化产物。
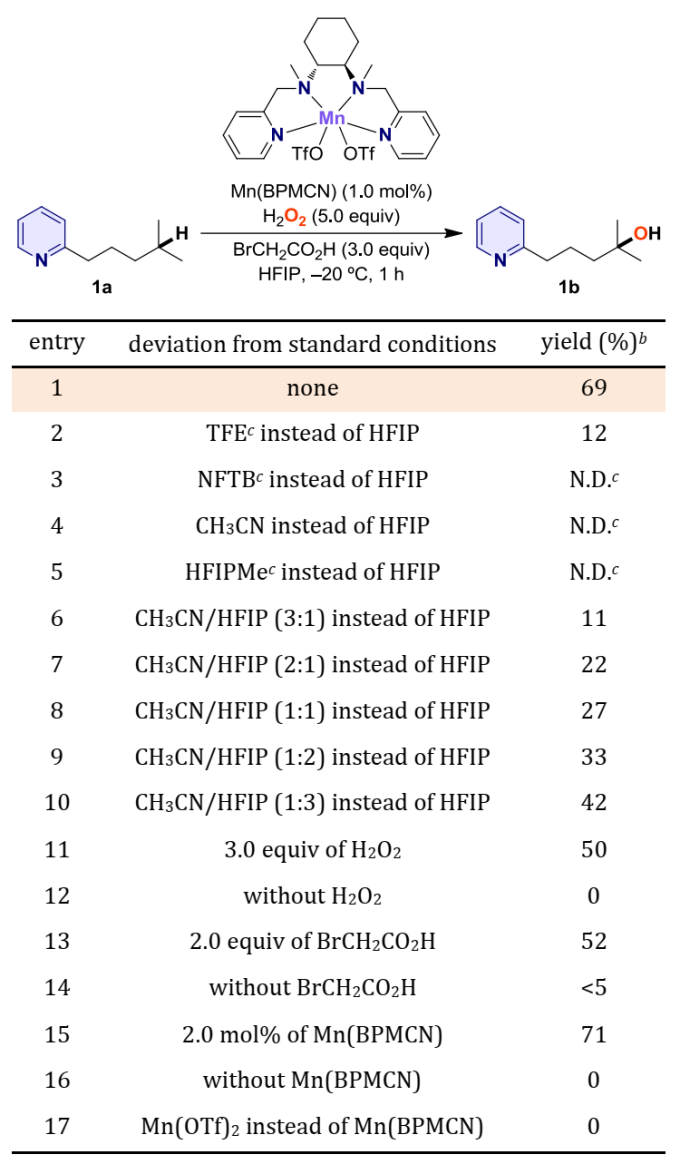
图2. 反应条件优化表格
作者在得到最优反应条件之后考察了对于六元氮杂芳环的耐受性(图3),结果显示单取代和二取代烷基吡啶、吡啶甲酸酯、烟酸酯、异烟酸酯和吡嗪酸酯均以良好的产率在远端三级C–H位点发生羟化。需要指出的是,对于相同含氮分子的氧化,本文使用1.0 mol%的Mn(BPMCN)作催化剂能够获得与White使用15 mol%的Fe(PDP)作催化剂的预保护策略相当甚至更高的产率。
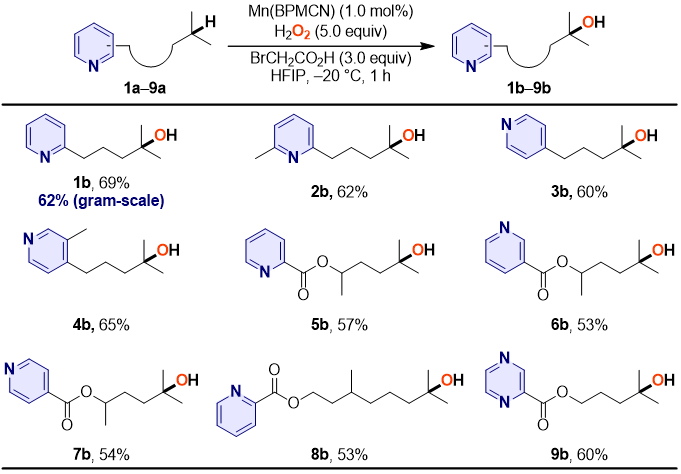
图3. 含六元氮杂环的分子中远端C‒H羟化
随后,作者考察了该催化体系对苯并氮杂芳环的兼容性(图4)。对于所有考察的底物都能以良好的产率获得远端C‒H羟化的叔醇产物,说明该催化体系能够耐受药学上重要的苯并氮杂芳环。
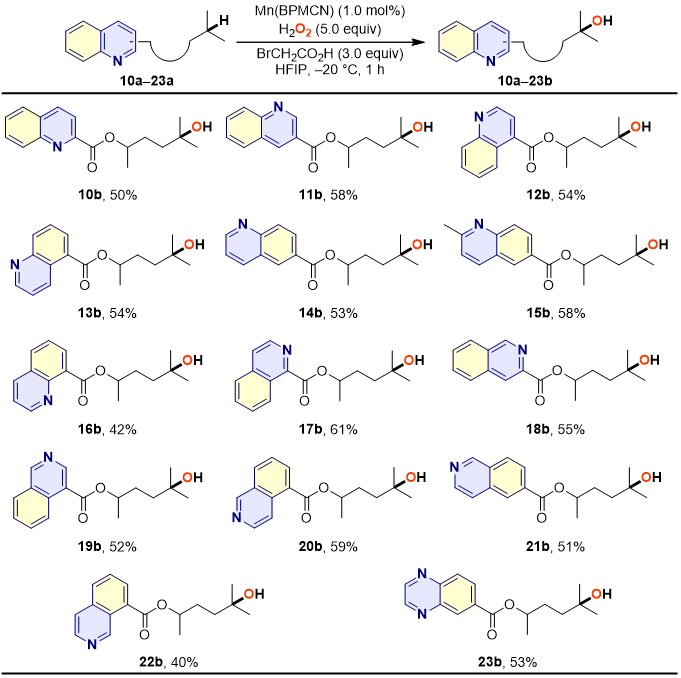
图4. 含苯并氮杂芳环的分子中远端C‒H羟化
另外,该反应还适用于含有五元氮杂芳环分子的远端C–H羟化反应(图5)。对于含有烷基侧链的异恶唑、噻唑、吡唑和1,3,4-恶二唑等底物,以较高的产率获得远端C–H氧化羟化产物。
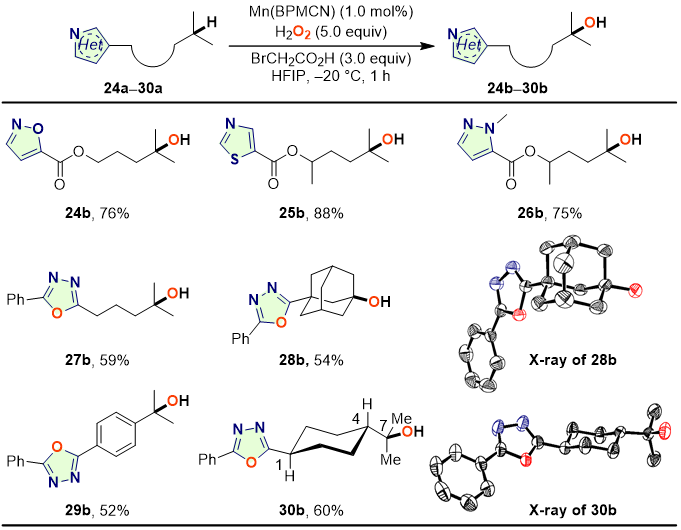
图5. 含五元氮杂芳环的分子中远端C‒H羟化
吡啶基甲醇、喹啉基甲醇和异喹啉基甲醇在药物、天然产物和生物活性分子中普遍存在,但这些同时含有碱性氮原子和仲醇的分子中非活化C–H的直接氧化官能化未见报道。因此,作者考察了同时含有氮杂芳环和羟基官能团分子的适用性(图6),研究结果发现既含有氮杂芳环又含有仲醇的分子能够直接进行远端C–H选择氧化,在温和的反应条件下从简单烷烃合成各种1,4-二醇,未观察到苄位醇羟基的氧化产物。
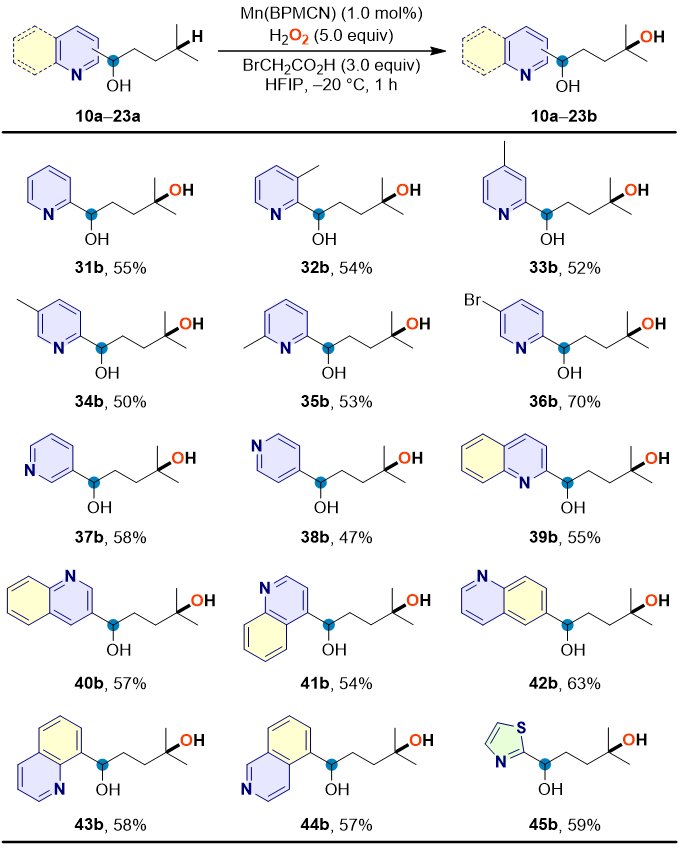
图6. 同时含有氮杂芳环和仲醇的分子中远端C‒H羟化
为证明HFIP与碱性氮原子之间存在强氢键相互作用,作者进行了系统的NMR研究(图7)。研究发现HFIP与底物1a混合后,HFIP的羟基信号出现了显著去屏蔽位移(图7C),同时,吡啶环上的4个质子的化学位移也发生了显著位移(图7C)。此外,作者还进行了1D NOE实验(图7D),通过探测空间上彼此接近的质子核之间偶极空间相互作用进一步确认底物1a和HFIP形成了强氢键。
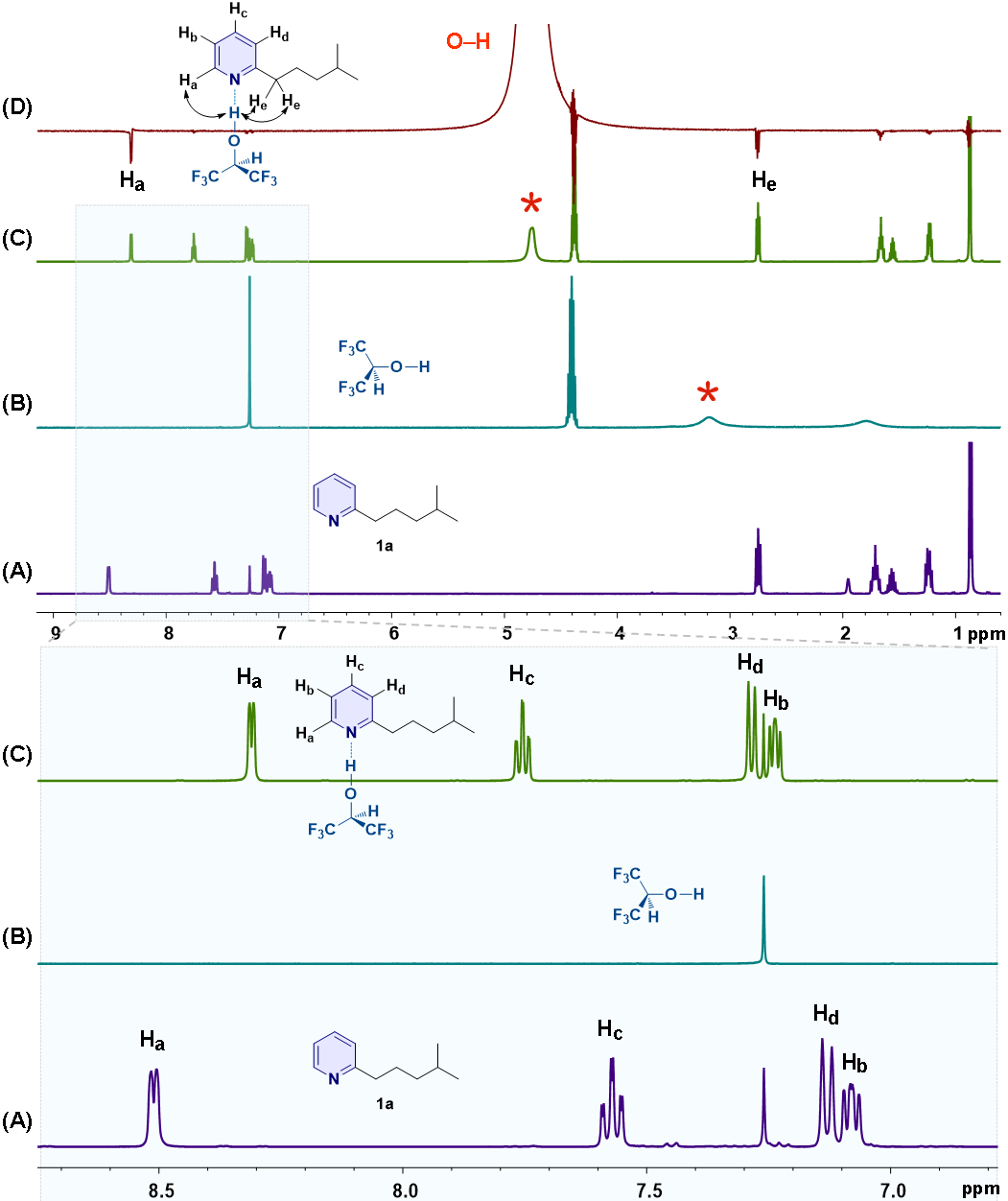
图7. 底物1a与HFIP之间存在强氢键
为进一步阐明HFIP在催化反应中所起的作用,作者进行了密度泛函理论计算(图8)。研究发现在HFIP存在下(图8B),MnV(O)向MnIII(κ2-OOC(O)CH2Br)的衰变和MnIII-OOH前体的可逆生成都得到了极大抑制,即在HFIP存在下,中间体MnV(O)(OC(O)CH2Br)的稳定性得到显著提高。另外,由于HFIP的氢键相互作用,MnV(O)(OC(O)CH2Br)在C–H羟化中的活性在氢原子摘取步骤中降低,而在氧反馈步骤中提高。
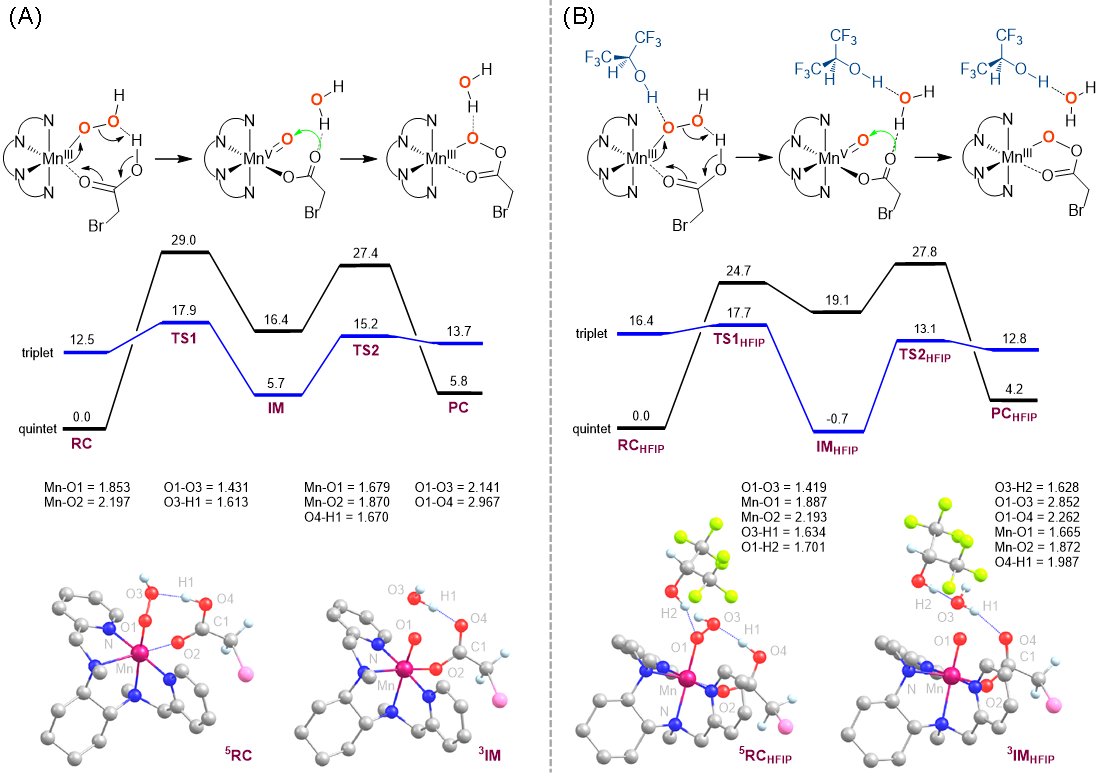
图8. 吉布斯自由能及在不存在(A)和存在(B)HFIP条件下MnIII-OOH经MnV(O)转化为MnIII(κ2-OOC(O)CH2Br)的关键结构信息
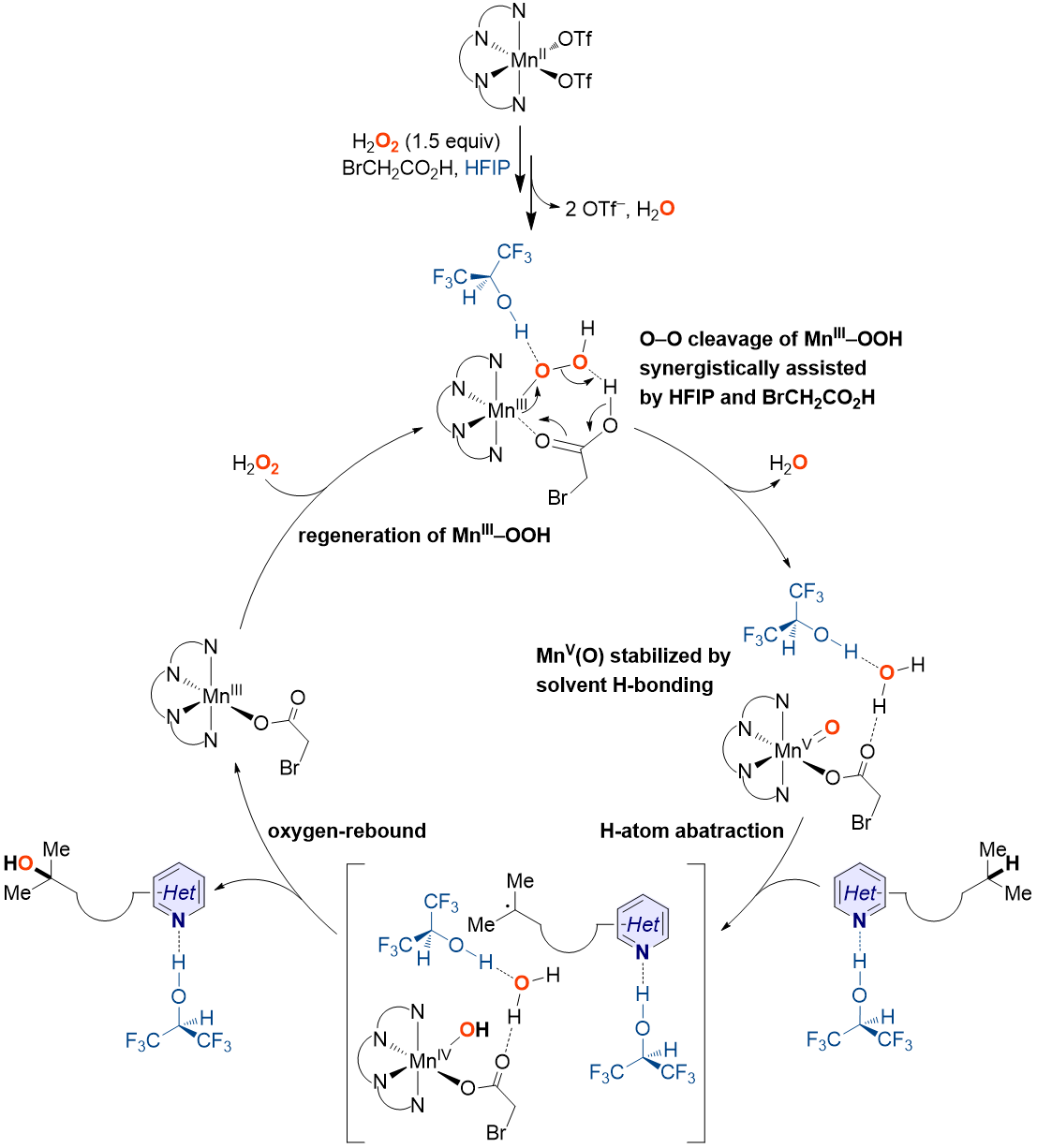
图9. 可能的反应机理
总 结
受金属氧化酶次级配位环境中的氢键作用启发,济南大学王斌课题组报道了一种溶剂氢键策略,实现了非血红素锰配合物催化以H2O2为氧源的含氮分子中远端C(sp3)–H氧化羟化。该方法的关键特征包括:(1)锰催化剂廉价易得且用量低;(2)无需强Lewis酸或Brønsted酸对碱性氮原子进行预保护;(3)优异的位点选择性。另外,作者还通过实验和理论方式对反应机理进行了研究(图9),阐明了HFIP在催化反应中所起的作用。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载























No comments yet.