
作者:杉杉
导读:
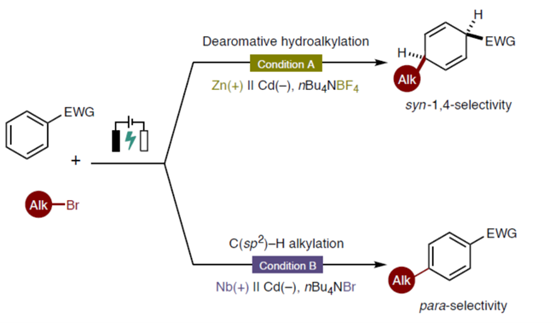
近日,哈尔滨工业大学(深圳)的夏吾炯等课题组在Nat. Chem.中发表论文,报道一种全新的有机电化学策略,实现了对缺电子芳烃和杂芳烃的去芳构化syn-1,4-氢烷基化反应,合成了一系列烷基化的syn-1,4-环己二烯衍生物,具有反应条件温和、操作简便、优异的化学/区域/立体选择性等优点。同时,通过对反应条件的调整,同样实现了(杂)芳烃的对位选择性C(sp2)–H烷基化反应。这两种反应均展现出广泛的底物适用范围,并与多种缺电子芳烃和烷基溴表现出优异的兼容性。
Dearomative syn-1,4-hydroalkylation and C(sp2)−H alkylation of arenes controlled by chemoselective electrolysis
C. Wan, C. Yang, M. Rueping, C. Zhu, L. Guo, W. Xia, Nat. Chem. 2025, ASAP. doi: 10.1038/s41557-025-02001-9.
正文:
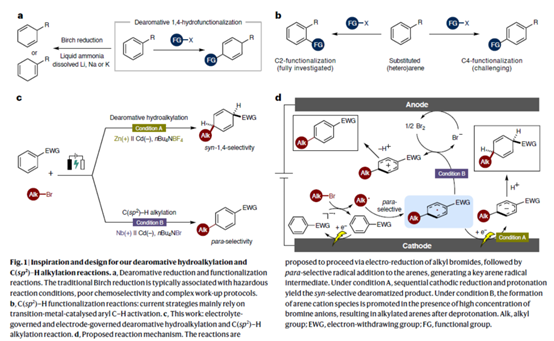
去芳构化反应是一类极为重要的策略,可将高度惰性的二维分子转化为三维结构。其中,Birch还原作为一种经典方法,但存在反应条件危险性高、化学选择性较差、后处理繁琐等弊端。随后,诸多研究团队开发了多种Birch还原的改良方法[1]。与简单的去芳构化还原不同,芳香环上官能团的引入与调控显著增加了去芳构化产物的复杂性,从而触及更富吸引力的化学空间(Fig. 1a)。然而,对于去芳构化官能团化反应的核心挑战在于克服芳香体系固有的稳定性。同时,在三维立体结构产物的形成过程中,对化学选择性、区域选择性与立体选择性的精准控制也具有难度。因此,将简单的苯环或杂环转化为官能团化的部分饱和环状骨架,仍具有挑战。另一方面,芳烃的C(sp2)–H键官能团化反应也是一类重要的策略,该反应能维持苯环的芳香性。其中,Friedel-Crafts反应是在芳环上引入烷基和酰基最直接的方法之一,然而该反应通常不适用于缺电子芳烃,且常伴随过度烷基化和异构化等副反应。近年来,诸多研究团队已开发出多种精巧的过渡金属催化策略,特别是在导向基团作用下实现邻位选择性C(sp2)–H官能团化的方法[2]。相比之下,实现远端对位C(sp2)–H官能团化仍面临重大挑战(Fig. 1b)。目前,对于远端C(sp2)–H烷基化反应方法通常一定的局限性[3],如使用过渡金属催化剂及配体、烷基化试剂适用范围受限、反应条件苛刻等。因此,开发通过可持续无过渡金属路径实现直接、高效且具有对位选择性的C(sp2)–H官能团化反应,具有至关重要的意义。受到电化学C(sp2)–H官能团化[4]相关研究报道的启发,这里,哈尔滨工业大学(深圳)的夏吾炯等课题组报道一种全新的有机电化学策略,实现了缺电子芳烃和杂芳烃的去芳构化syn-1,4-氢烷基化反应。同时,通过对反应条件的修改,同样实现了(杂)芳烃的对位选择性C(sp2)–H烷基化反应(Fig. 1c)。从机理上讲,首先发生烷基溴化物还原,随后生成的自由基对芳烃进行对位选择性加成,形成关键的芳基自由基中间体。在Condition A中,经历连续阴极还原与质子化后得到去芳构化产物。在Condition B中,在高浓度溴离子存在时,反应促进芳烃阳离子物种生成,经去质子化后最终获得烷基化芳烃(Fig. 1d)。
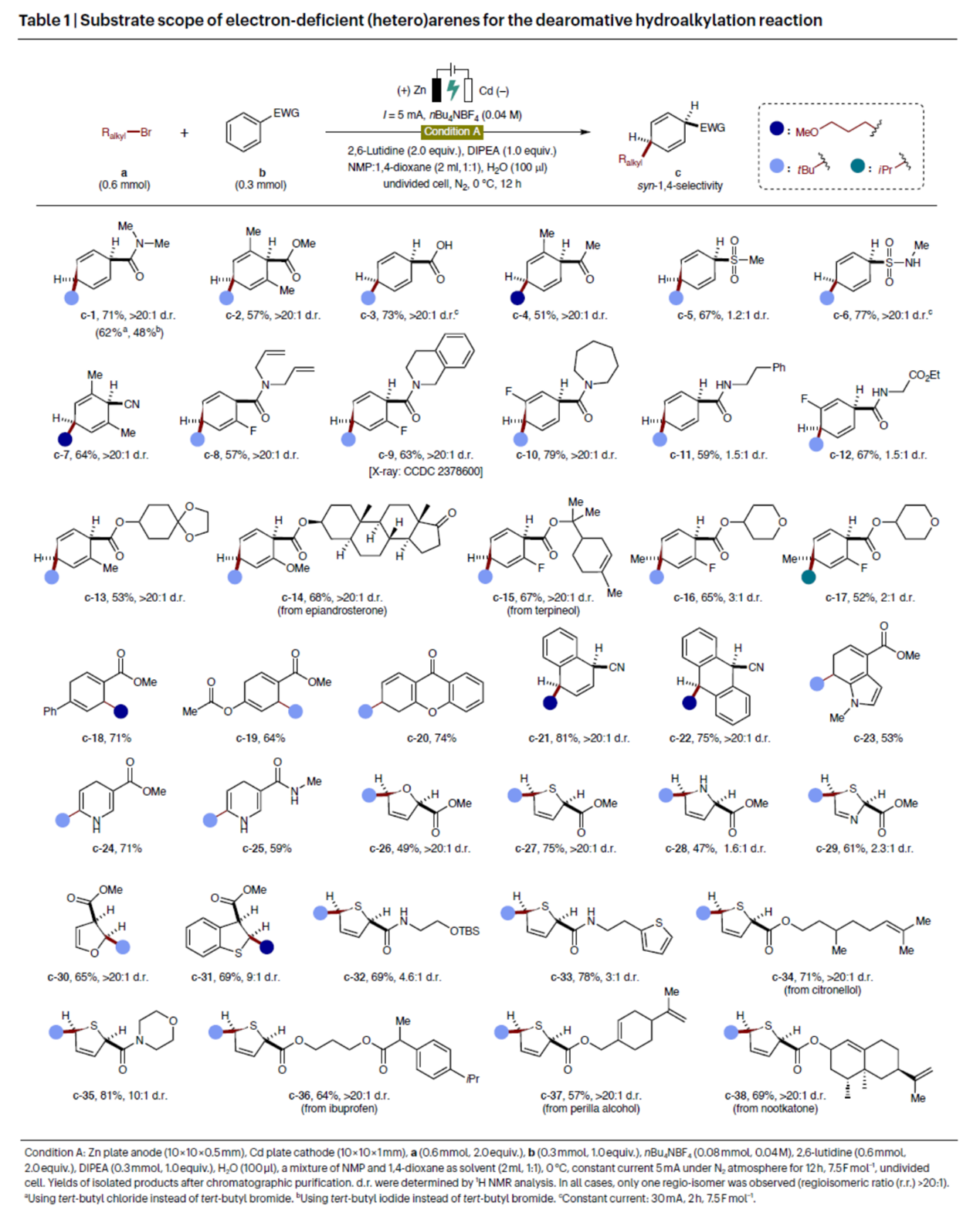
首先,作者进行相关反应条件的优化筛选(Supplementary Table 1)。进而确定了去芳构化syn-1,4-氢烷基化反应最佳的反应条件为(即Condition A):采用锌板作为阳极,镉板作为阴极,电流为5 mA,nBu4NBF4作为电解质,2,6-二甲基吡啶作为碱,DIPEA/H2O作为添加剂,在NMP/1,4-二氧六环(1:1)混合反应溶剂中,反应温度为0 oC,最终获得71%分离收率的去芳构化氢烷基化产物c-1(>20:1 dr)。同时,对位选择性C(sp2)–H烷基化反应最佳的反应条件为(即Condition B):采用铌板作为阳极,镉板作为阴极,电流为5 mA,nBu4NBr作为电解质,2,6-二甲基吡啶作为碱,在NMP/1,4-二氧六环(1:1)混合反应溶剂中,反应温度为0 oC,最终获得57%分离收率的对位选择性C(sp2)–H烷基化产物d-1。
在上述的最佳反应条件下,作者对去芳构化氢烷基化反应中的缺电子(杂)芳烃的底物范围进行了扩展(Table 1)。首先,芳烃上含有简单的酰胺基、烷氧羰基、羧基、羰基、磺酰胺基和腈基取代时,均可顺利进行反应,获得相应的产物c-1–c-7,收率为51-77%,dr > 20:1。一系列复杂取代的芳香酰胺,也能够进行反应,获得相应的产物c-8–c-12,收率为57-79%,dr为1.5:1->20:1。同时,一系列含有复杂烷氧羰基取代的芳烃,也能够进行反应,获得相应的产物c-13–c-17,收率为52-68%,dr为2:1->20:1。然而,当对位存在强供电子基团(如-OAc基团)或苯基时,去芳构化氢烷基化反应的烷基化位点将转移至吸电子基团的邻位(如c-18与c-19)。其次,多种稠合芳烃,也与体系兼容,获得相应的产物c-20–c-23,收率为53-81%。此外,一系列杂芳烃底物,如烟酸甲酯、N-甲基烟酰胺、呋喃、噻吩、吡咯、噻唑等,均与体系兼容,获得相应的产物c-24–c-38,收率为47-81%。
其次,作者对去芳构化氢烷基化反应中烷基溴的底物范围进行了扩展(Table 2)。首先,环状和非环状三级烷基溴,均可顺利进行反应,获得相应的产物c-39–c-45,收率为54-78%,dr为2:1->20:1。其次,环状和非环状二级烷基溴,也是合适的底物,获得相应的产物c-46–c-53,收率为48-77%,dr为1.2:1->20:1。此外,各种官能团化的一级烷基溴,也能够顺利进行反应,获得相应的产物c-54–c-62,收率为47-78%,dr为1:1->20:1。
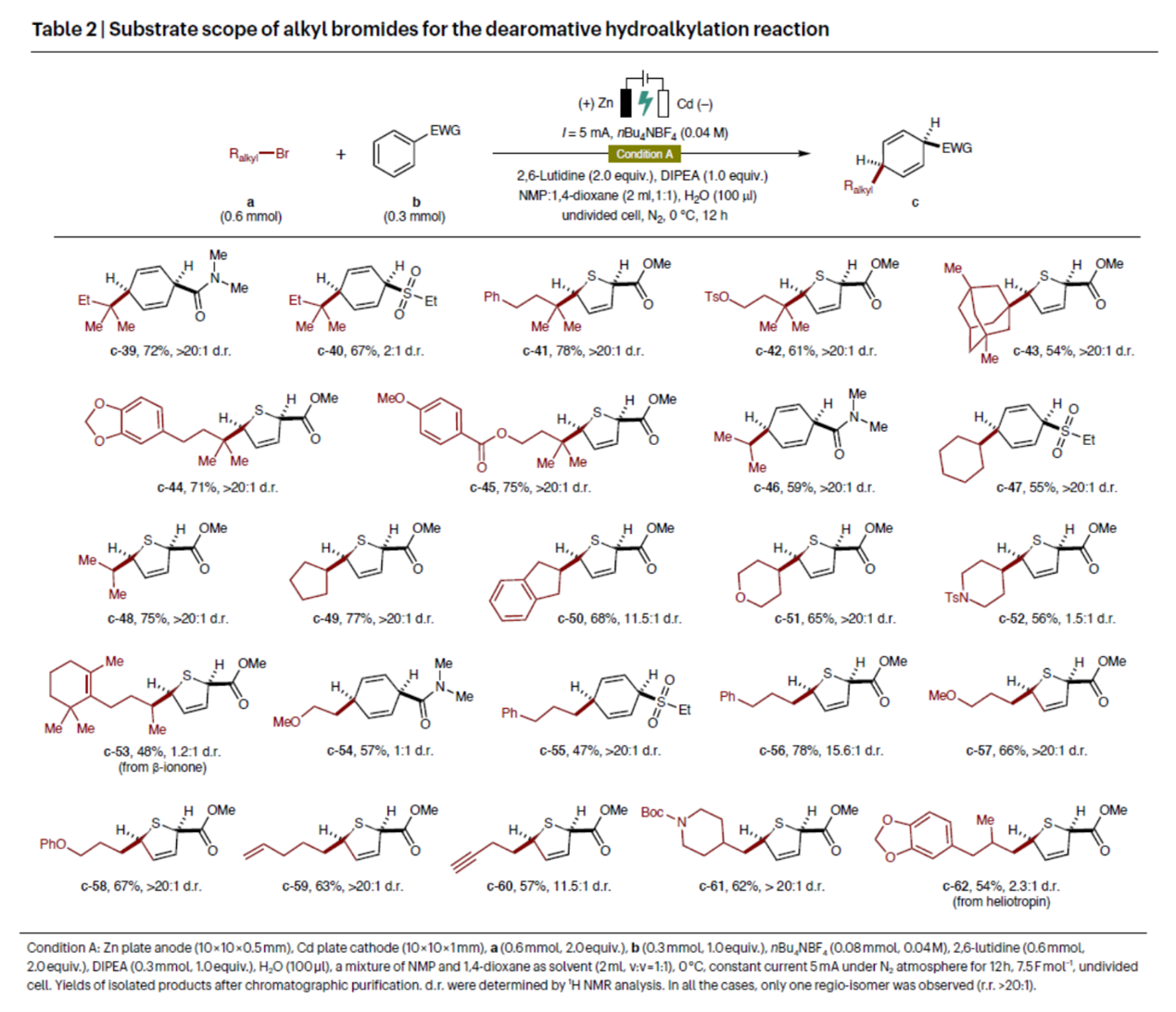
紧接着,作者对对位选择性C(sp2)–H烷基化反应的底物范围进行了扩展(Table 3)。首先,一系列不同电性取代的芳烃,均可与叔丁基溴顺利进行反应,获得相应的产物d-1–d-32,收率为30-81%。其次,一系列一级/二级/三级烷基溴衍生物,均可与芳基乙酰胺进行反应,获得相应的产物d-33–d-58,收率为41-74%。
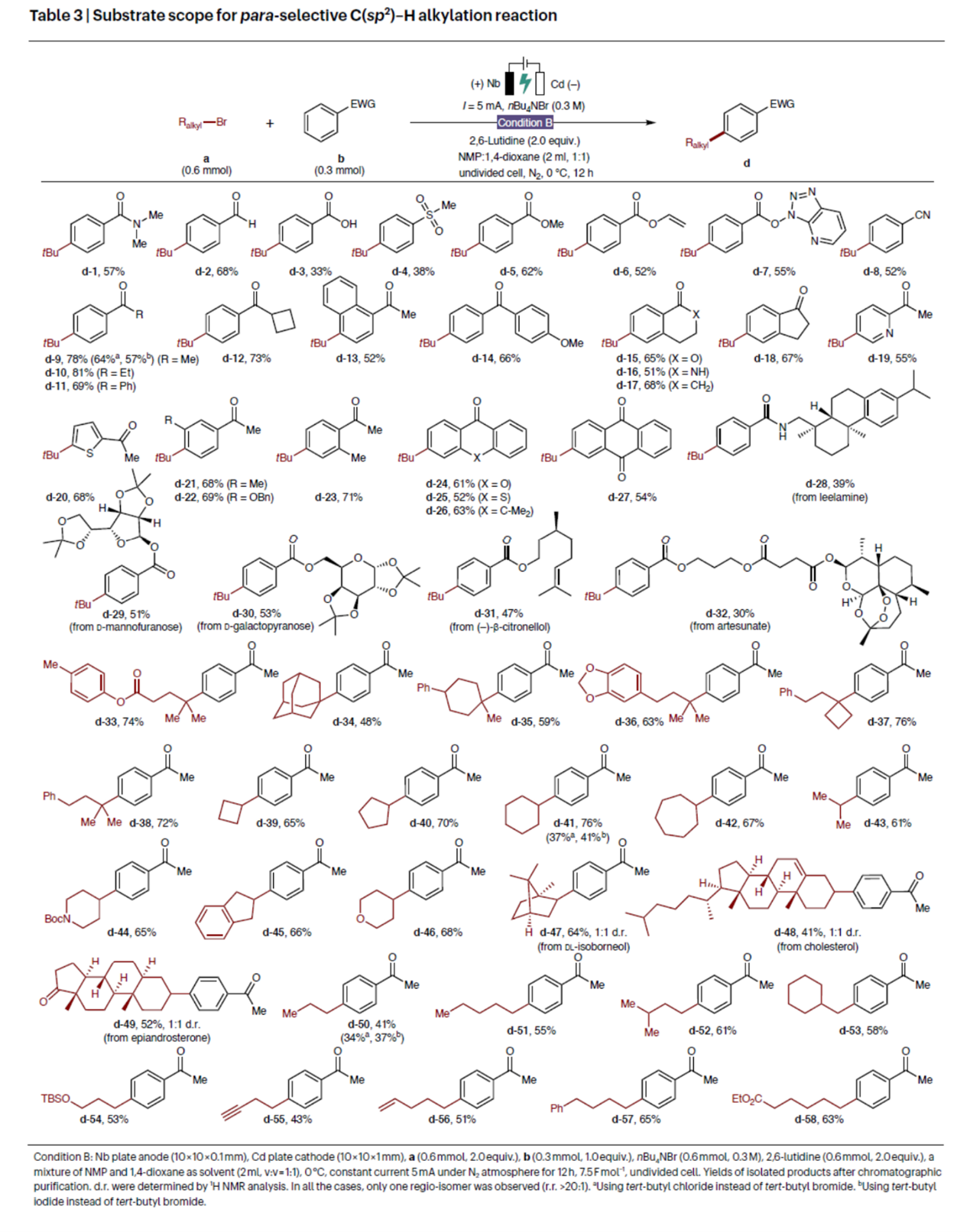
随后,作者对反应的实用性进行了研究(Fig. 2)。首先,a-1与b-1的去芳构化氢烷基化反应的克级规模实验,同样可以64%的收率得到产物c-1。同时,在a-1与b-1的对位选择性C(sp2)–H烷基化反应的克级规模实验中,用快速且可扩展的连续流工艺,同样可以62%的收率得到产物d-9。其次,以3,5-二甲氧基苯甲酸甲酯(e-1)与1-溴丙烷为底物,通过上述的对位选择性C(sp2)–H烷基化反应(制备e-2)与去芳构化氢烷基化反应,可以两步38%的总收率得到化合物e-3,其是制备Atiprimod的前体。类似的,以2-甲氧基苯甲酸酯(e-6)与2-溴丙烷为底物,通过上述的对位选择性C(sp2)–H烷基化反应,可以71%的收率获得化合物e-5,其是合成(±)-Periplanone C的前体,避免了前期文献中步骤长、反应时间长等弊端。同时,以吡啶甲酸甲酯(e-8)与1-溴丁烷为底物,通过上述的对位选择性C(sp2)–H烷基化反应,可以51%的收率获得化合物e-9,其是合成富马酸的前体,避免了前期多步反应过程。
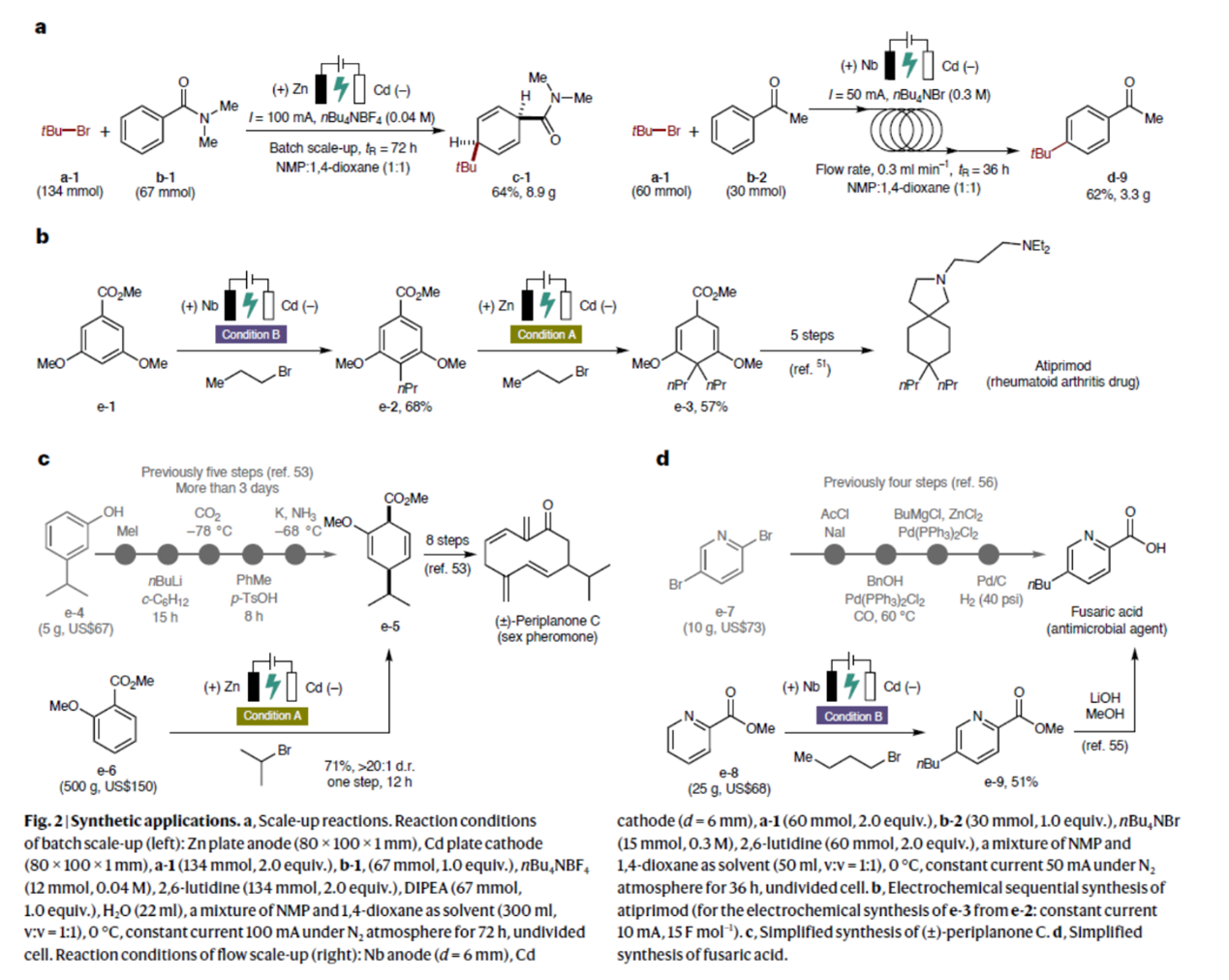
最后,作者对反应的机理进行进一步研究(Fig. 3)。首先,CV实验结果表明,b-1在阴极表面优先被还原,从而促进烷基溴的后续还原反应,最终生成去芳构化产物(Fig. 3a与Fig. 3b)。其次,对照实验结果表明,通过添加nBu4NBr作为支持电解质,实现了高化学选择性(Fig. 3c)。自由基实验结果表明,该反应可能涉及烷基溴经电化学还原生成关键烷基自由基物种的过程(Fig. 3d与Fig. 3e)。同位素标记实验结果表明,水作为主要的氢源(Fig. 3f)。此外,作者将该电化学策略的合成应用拓展至Birch型还原:在Condition A下,以水为氢源,成功将缺电子杂芳烃(b-3)转化为Birch型去芳构化产物e-15,收率达67%(Fig. 3g)。
总结:
哈尔滨工业大学(深圳)的夏吾炯等课题组报道一种全新的有机电化学策略,实现了对缺电子芳烃和杂芳烃的去芳构化syn-1,4-氢烷基化反应,合成了一系列烷基化的syn-1,4-环己二烯衍生物。同时,通过对反应条件的修改,同样实现了(杂)芳烃的对位选择性C(sp2)–H烷基化反应。此外,上述两种策略具有温和的反应条件、广泛的底物范围、出色的官能团兼容性以及优异的化学/区域/立体选择性等优势。
参考文献:
- [1] A. Chatterjee, B. Konig, Angew. Chem. Int. Ed. 2019, 58, 14289. doi:10.1002/anie.201905485.
- [2] C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes, M. Schnürch, Chem. Soc. Rev. 2018, 47, 6603. doi:10.1039/C8CS00201K.
- [3] Z. Jiao, L. H. Lim, H. Hirao, J. S. Zhou, Angew. Chem. Int. Ed. 2018, 57, 6294. doi:10.1002/anie.201801967.
- [4] Z. Zhao, R. Zhang, Y. Liu, Z. Zhu, Q. Wang, Y. Qiu, Nat. Commun. 2024, 14, 3823. doi:10.1038/s41467-024-48262-9.
















No comments yet.