

本文作者:杉杉
导读
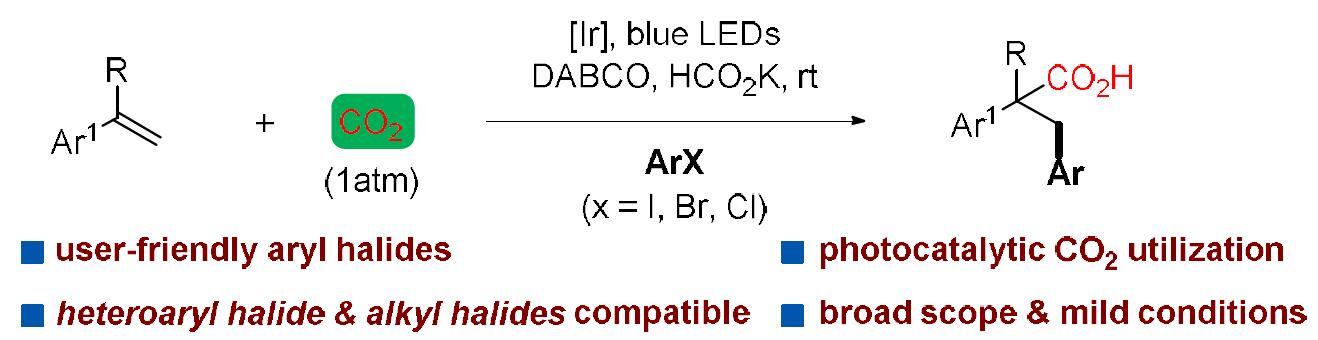
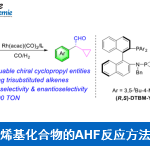
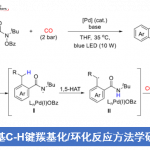
近日,中科院(福建物质结构研究所)李纲教授课题组在美国化学学会杂志(Journal of the American Chemical Society)发表论文,首次报道了在可见光促进下,实现苯乙烯化合物与CO2、芳基卤化物的区域选择性还原羰基化反应。该反应具有广泛的底物范围,如吡啶基卤化物、烷基卤化物、芳基碘化物、溴化物,甚至氯化物均与反应体系兼容。该策略使用芳基卤化物作为自由基源,二氧化碳作为羰基源,通过光催化,实现Meerwein芳基化反应。
Visible-Light-Driven Reductive Carboarylation of Styrenes with CO2 and Aryl Halides
Hao Wang, Yuzhen Gao, Chunlin Zhou, and Gang Li
J. Am. Chem. Soc. ASAP DOI: 10.1021/jacs.0c03144
正文
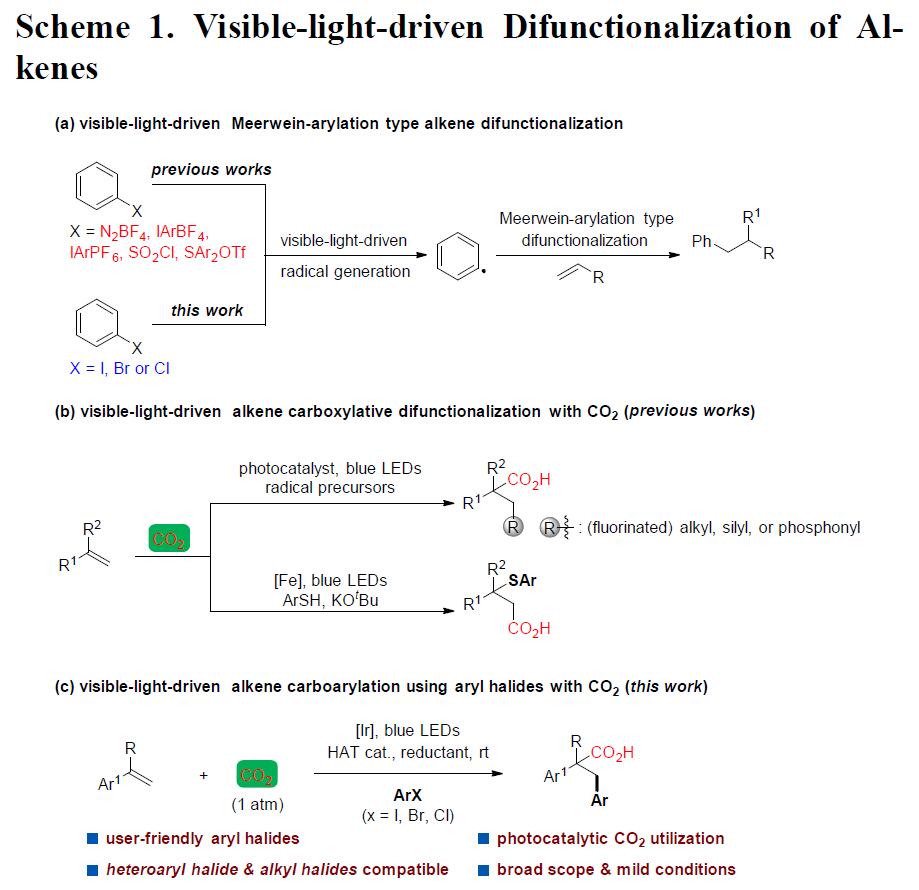
在温和的反应条件下,通过可见光促进的光氧化还原催化反应(PRC),作为合成复杂有机分子的有效方法。此外,烯烃的双官能化反应也是高效合成复杂性分子的重要方式,除了过渡金属催化或其他自由基介导以外,PRC也作为一种高效的途径。在众多的反应中,可见光促进烯烃双官能化反应最为常见,通常以芳基重氮盐、二芳基碘鎓盐、芳基磺酰氯等作为芳基自由基的来源(Scheme 1a)。然而,迄今为止,一些更为稳定、廉价且易获得的芳基卤化物尚未应用于此体系中,可能是由于可见光很难从芳基卤化物生成高反应性的芳基,因为与芳基重氮盐、二芳基碘鎓盐或芳基磺酰氯相比,芳基卤化物需要更高的负还原电位。
近年来,CO2作为一种可持续、丰富、低成本、无毒的原料,一直作为化学合成领域的研究热点,而利用光催化,可以很好将其引入反应体系中。值得注意的是,仅有少量文献已报道关于使用中性光催化剂,实现CO2与烯烃的双官能化反应,获得一些具有价值的羧酸衍生物(Scheme 1b)。此外,其他课题组也报道了使用CO2与各种自由基前体反应,经可见光诱导,实现区域选择性的碳羧化、硅烷基化、膦酰基羧化、烯烃的硫代羧化等过程。然而,这些光催化体系均不适用于CO2、芳基卤化物、烯烃的三组分芳基化反应,因为要使芳基卤化物参与此类反应,必须使用一种新颖的高度还原的催化剂。基于这一挑战,尚未有文献报道关于通过可见光促进烯烃、CO2和芳基卤化物的Meerwein芳基化羰基化反应。在此,中科院(福建物质结构研究所)李纲教授课题组首次提出使用可见光作为催化剂,使用廉价的HCO2K作为末端还原剂,可实现苯乙烯、CO2、芳基卤化物的高度区域选择性的还羰基化反应,获得多种具有价值的氢化肉桂酸衍生物 。此外,该芳基卤化物的范围可进一步扩展到吡啶基卤化物、烷基卤化物甚至芳基氯化物等(Scheme 1c)。
首先,作者以1,1-二苯基乙烯、碘苯和CO2作为模型底物,进行了相关羰基化反应条件的筛选(Table 1)。 经大量反应条件筛选之后,当以DABCO作为氢原子转移催化剂,[Ir(ppy)2(dtbbpy)]PF6作为光催化剂,HCO2K作为末端还原剂,K2CO3作为碱,可在DMSO溶剂中,获得78%收率的目标产物1(entry 1)。此外,为了便于分析和分离产物,作者将原始的羧酸产物转化为甲酯。当不使用光催化剂或在无光的条件下,均未检测到产物,从而表明反应需由光诱导(entries 2-3)。而在氮气气体中反应时,收率略有下降,表明HCO2K的氧化或K2CO3的酸化也可能形成CO2(entry 4)。在不存在末端还原剂HCO2K时,则无法获得任何产物(entry 5)。当使用2.5当量的DABCO,但体系中不存在HCO2K,只能形成痕量的产物(entry 6)。而对其它HAT催化剂筛选后,DABCO具有较高的催化活性(entries 7-10)。这些结果可能表明,HCO2K/DABCO的结合作为该还原羰基化反应中有效的电子供体。此外,K2CO3对于该反应仅稍有益处(entry 11),而其他碱(如Na2CO3和Cs2CO3)也同样有效(entries 12-13)。最后,作者也对溶剂进行筛选(DMF、DMA),发现DMSO作为最佳溶剂(entries 14-15)。

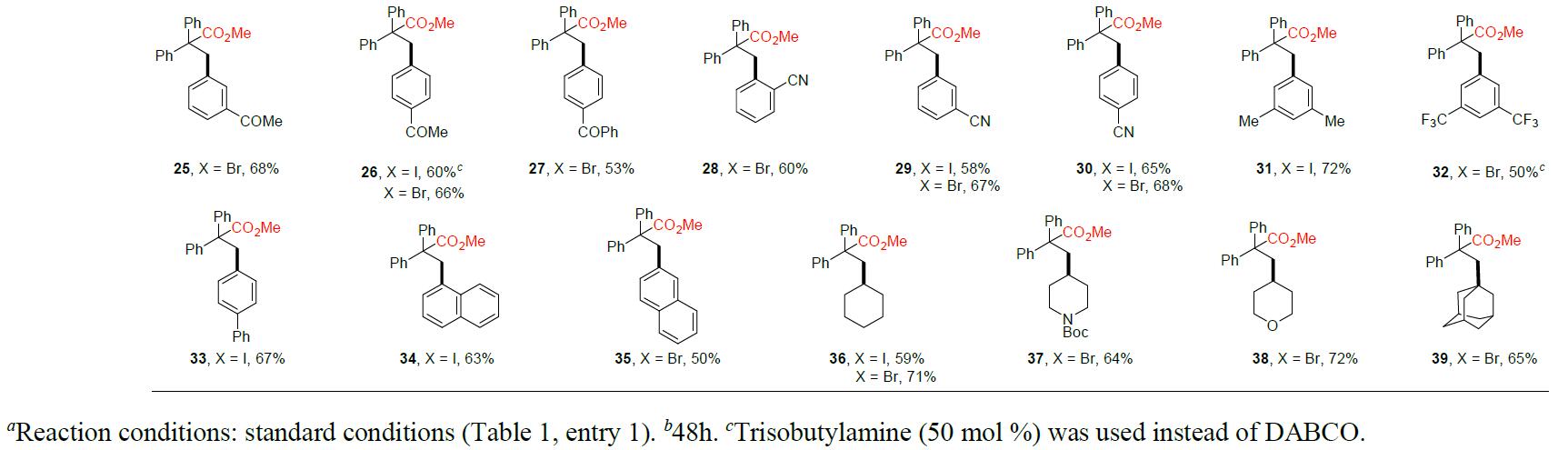
在获得上述最佳反应条件后,作者固定1,1-二苯乙烯,开始对各种芳基碘化物和溴化物以及一些具有代表性的烷基卤化物进行了扩展(Table 2)。首先,使用廉价的溴苯仅比碘苯获得产物1的收率稍差些。值得注意的是,在该反应体系中不受电子效应的影响,均可获得相应的目标产物2–33,如给电子基(甲基、甲氧基、甲硫基等),吸电子基(氟、氯代、硼酸酯、三氟甲基、羰基、氰基等)。但是,一些带有给电子基团的底物(如产物2–5的卤化物),芳基碘化物的收率高于芳基溴化物。同样,对于某些具有吸电子基团的底物,如产物25、28和32的溴化物,芳基溴化物的收率高于芳基碘化物。而且,邻位取代的芳基卤化物通常比间位和对位取代的的产物收率偏低,如产物5–7、12–14和28–30。此外,碘代萘(34)和溴代萘(35)也与体系兼容。值得注意的是,带有氯或硼酸酯基团的芳基碘化物/溴化物底物也可与体系兼容,获得相应的产物15–18,为进一步衍生化提供了多种可能。随后,作者开始对烷基卤化物的可行性进行了研究。令人满意的是,代表性的仲烷基卤化物产物36–38,可在优化的反应条件下顺利进行。此外,叔丁基溴化物(39)也可用于此反应。

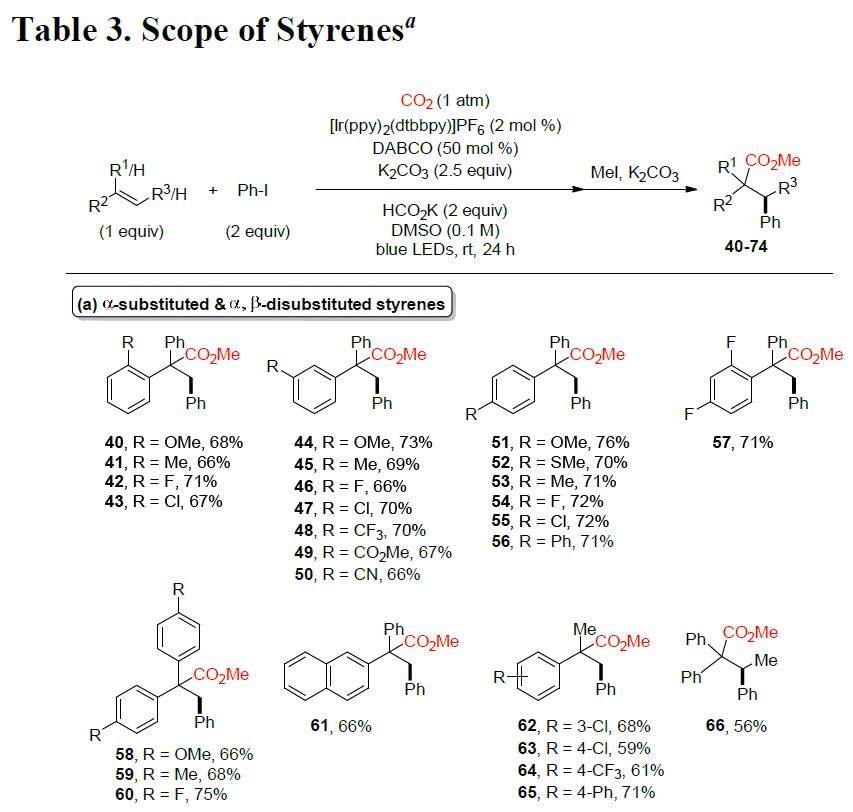
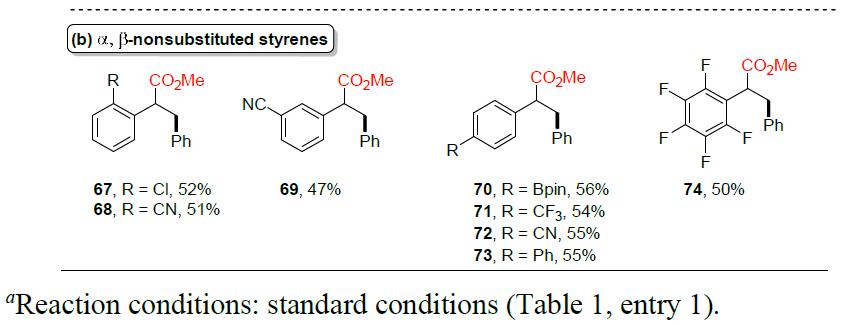
随后,作者对苯乙烯的底物范围进行了相关的扩展(Table 3)。反应结果表明,该反应不受电子效应和定位效应的影响,均可获得相应α-取代产物40–65。同时,位阻较大的α,β-二取代苯乙烯底物也可以中等收率获得相应的产物66。但是,α,β-非取代苯乙烯衍生物,仅获得中等收率的产物67–74。值得注意的是,硼酸酯基团(70)也可保持完整的构型,为后期修饰提供了多种可能。然而,脂族烯烃和环状苯乙烯,均与该体系不兼容。
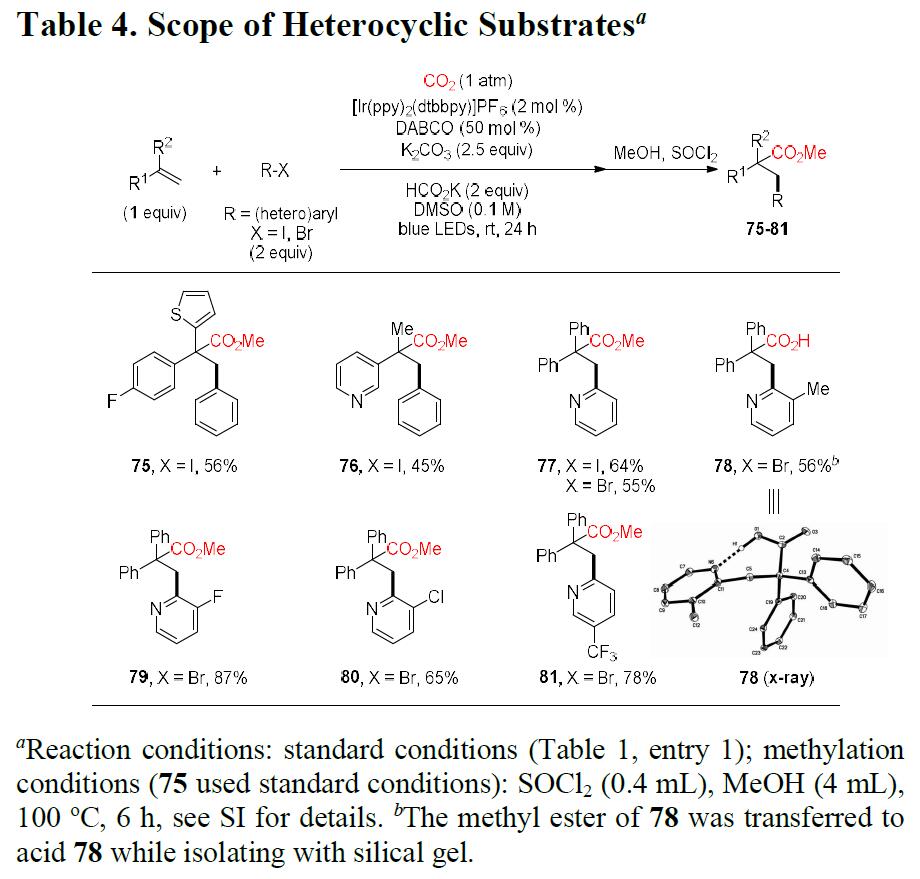
紧接着,作者将底物范围扩展到更具挑战性的缺电子的吡啶基卤化物(Table 4)。反应结果表明,具有硫代呋喃基或吡啶基的烯烃均可以中等收率获得所需的产物75和76。值得注意的是,该反应还可与其它的吡啶基卤化物底物反应,从而以良好的收率获得相应的产物77–81。此外,通过产物78的单晶X-射线结构分析进一步证明了结构的正确性。
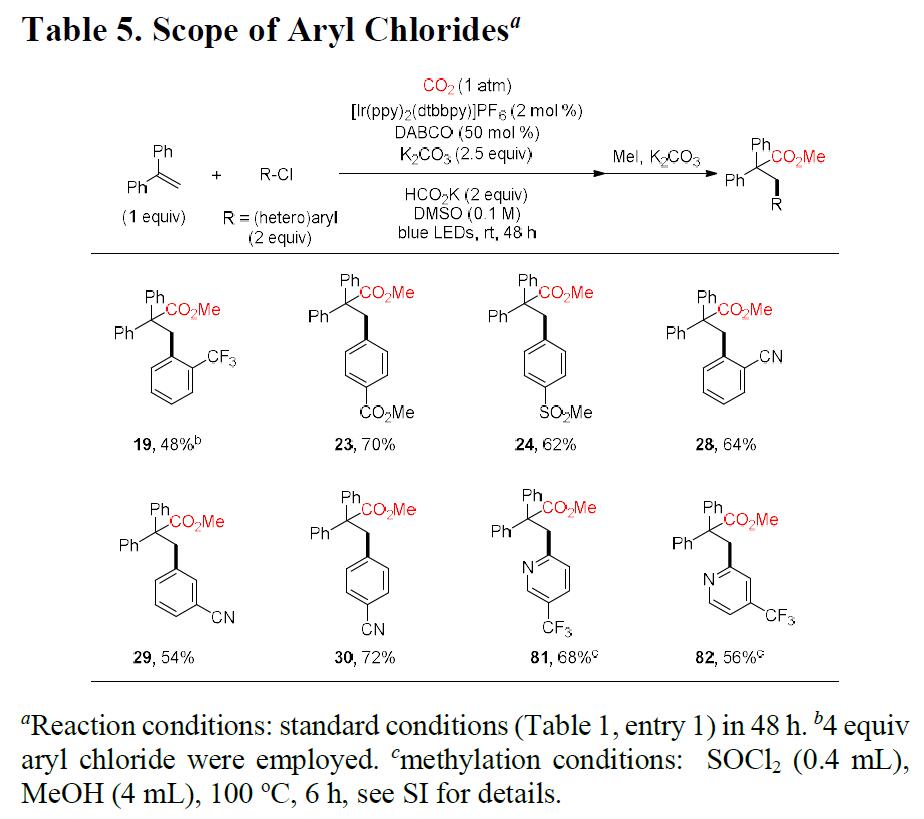
为了进一步证明该反应的底物范围的广泛性,作者选择使用反应活性更低的芳基氯化物(Table 5)。反应结果表明,一些更为廉价的缺电子的(杂)芳基氯化物均可发生羰基化反应,从而获得相应的产物。
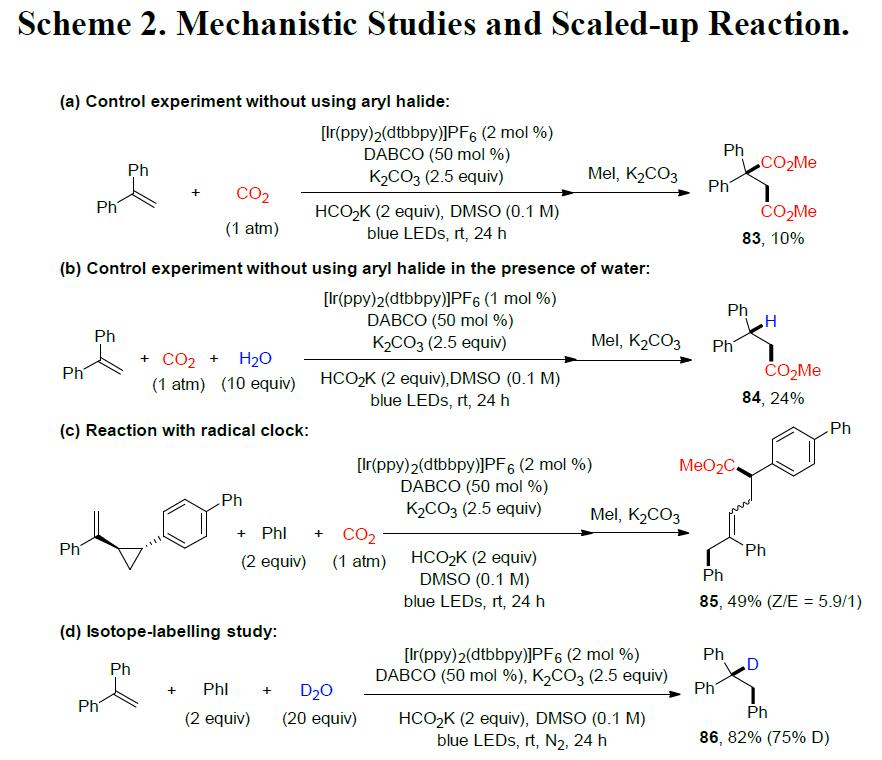
为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 2)。首先,在标准条件下,体系不存在芳基卤化物时,获得羧基化产物83和84,从而表明在该反应条件下可能会生成CO2自由基阴离子(Scheme 2a, 2b)。当进行自由基钟实验时,获得开环产物85,表明该反应可能生成苄基自由基(Scheme 2c)。而通过同位素标记实验和使用醛作为亲电试剂的对照实验(Scheme 2d, 2e),间接表明可能存在苄基阴离子中间体。此外,为了确定产物的羧基来源,作者使用13CO2(99%13C)气体,最终在产物的羧基中发现74%13C(Scheme 2f)。最后,该反应可以轻松扩大至5 mmol,而产物的收率没有明显降低(Scheme 2g)。
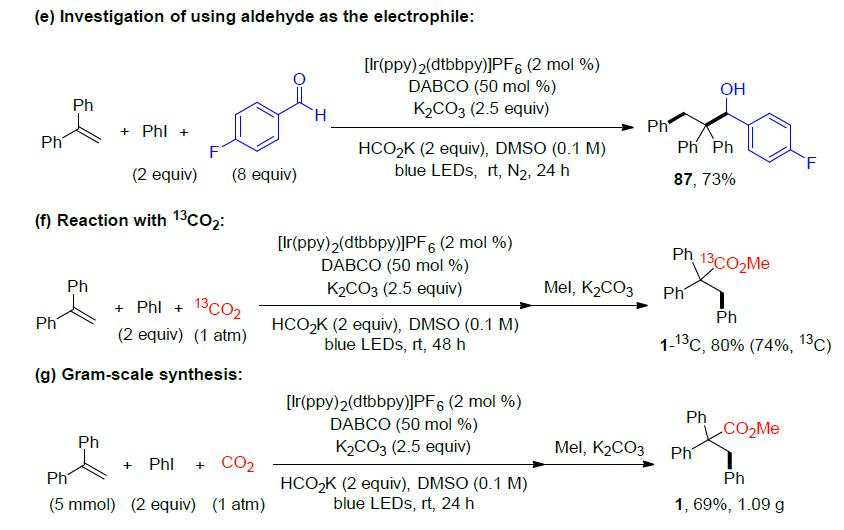
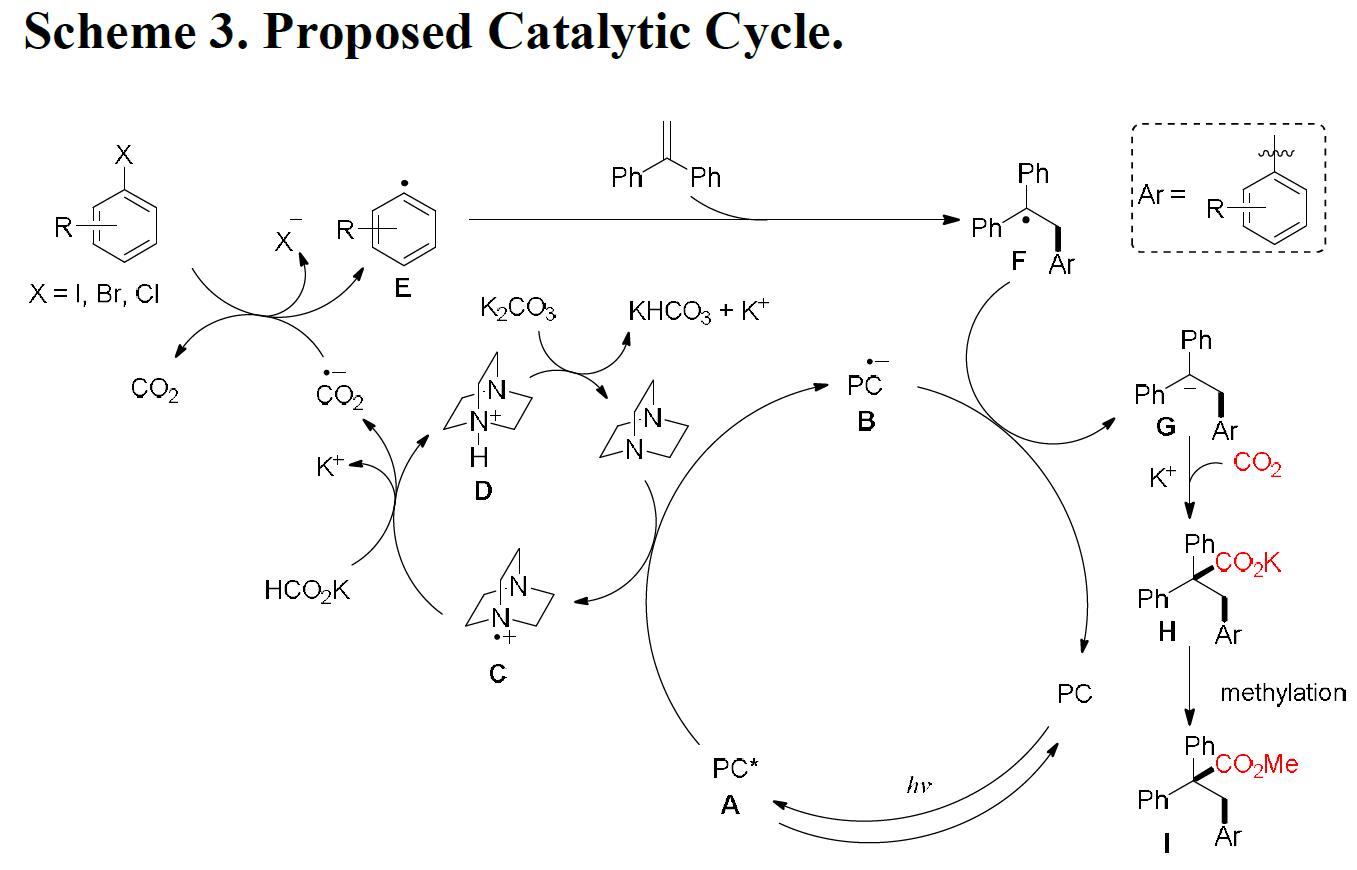
根据上述的实验和相关文献的查阅,作者提出了一种可能的反应机理(Scheme 3)。首先,PC在蓝光照射下,在产生激发A,经DABCO还原淬灭,得到还原的B和DABCO的自由基阳离子C。自由基阳离子C与阳离子D共同促进氢原子从HCO2K中转移,从而获得CO2自由基阴离子。CO2自由基阴离子具有很高的还原电位,通常可通过0.1-0.6 V的过电位进行补充。因此,芳基卤化物可被CO2自由基阴离子还原,形成芳基自由基E,其经区域选择性加成至烯烃上获得苄基自由基F。紧接着,经还原的光催化剂B的单电子转移过程形成苄基阴离子中间体G。然后,G再与CO2发生亲核加成反应,生成羧酸盐H,甲基化后将其转化为甲酯。应当指出,目前尚不能排除苄基中间体可以与CO2可逆反应,然后经还原的B到羧基中间体后再进行SET,从而获得羧酸盐H。
总结
中科院(福建物质结构研究所)李纲教授课题组报道了第一个可见光催化,实现苯乙烯衍生物、CO2和芳基卤化物的区域选择性羰基化的反应例子,获得多种具有有价值的肉桂酸衍生物。该反应具有广泛的底物范围,包括各种芳基卤化物、芳基氯化物、吡啶基卤化物、烷基卤化物等。该策略使用芳基卤化物作为自由基源,二氧化碳作为羰基源,通过光催化,实现Meerwein芳基化反应。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.