
导读:
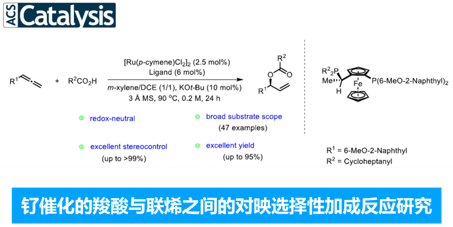
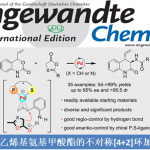
近日, 德国Albert-Ludwigs大学有机化学研究所 (Institut für Organische Chemie, Albert-Ludwigs-Universität)的B. Breit课题组在ACS Catal.中发表论文,报道一种钌催化的羧酸与联烯之间的加成反应方法学,进而成功完成一系列具有支链的烯丙基酯分子的构建。接下来,作者基于Josiphos配体骨架,成功设计出全新的手性配体,进而以优良的反应收率 (高达95%)与对映选择性 (高达99% ee),获得相应的手性烯丙基酯产物。之后,该小组通过氘标记实验研究,进而提出合理的反应机理。此外,研究发现,通过上述策略获得的支链烯丙基酯产物能够进一步转化为一系列重要的五元与六元环对映纯内酯分子。
Ruthenium-Catalyzed Enantioselective Addition of Carboxylic Acids to Allenes
J. Hu, F.Bauer, B. Breit, ACS Catal. 2021, 11, 12301. doi:10.1021/acscatal.1c03306.
正文:
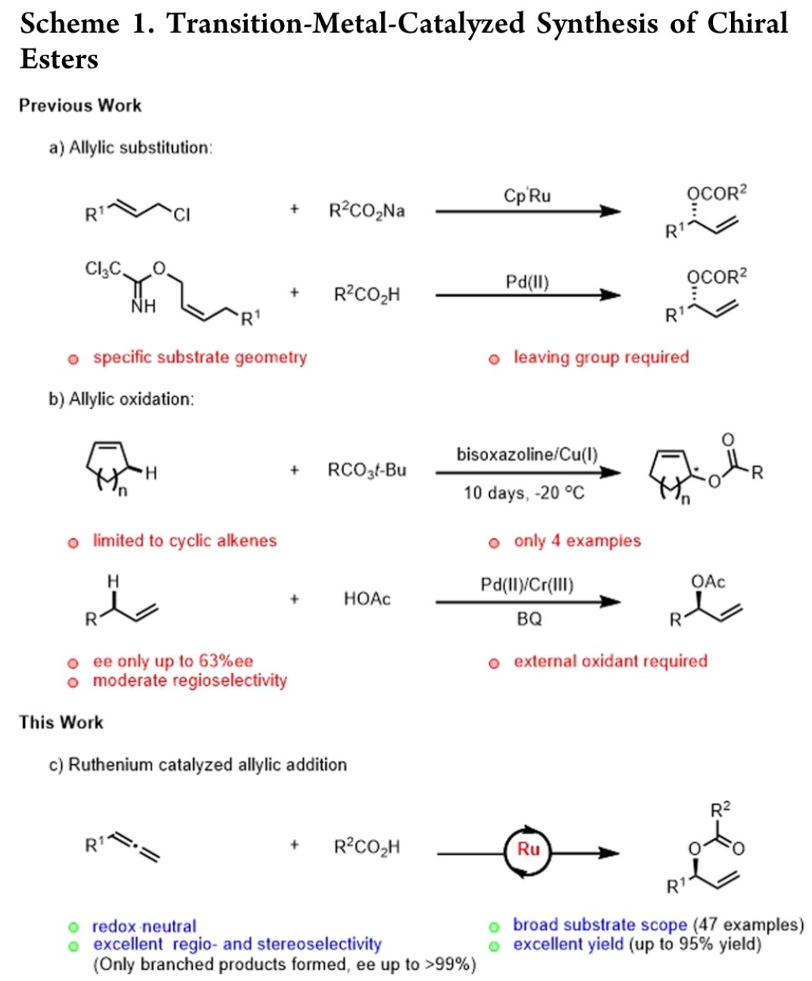
过渡金属钌由于具有多种不同的氧化态以及成本低廉等优势,在一系列有机合成转化方法学的研究中备受广泛关注[1]。同时,近年来,对于过渡金属催化的手性烯丙酯类分子的对映选择性合成方法学研究,已经取得诸多进展 (Scheme 1a-1b)[2]-[3]。然而,上述策略中,底物应用范围极为有限,同时,难以实现优良的对映选择性控制。
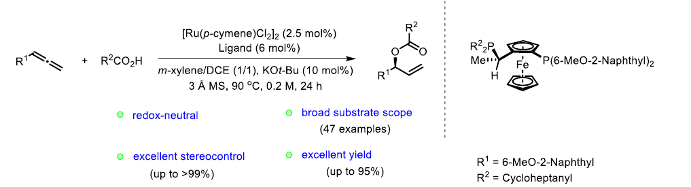
之后,研究发现,采用羧酸与联烯分子之间进行的不对称烯丙基加成反应方法学,能够有效地克服上述反应策略中存在的诸多局限,同时,能够获得优良的原子经济性。这里,受到本课题组前期对于联烯或炔基化合物参与的烯丙基加成反应方法学[4]研究的启发,作者成功设计出一种钌催化的羧酸与联烯之间的加成反应方法学,进而成功实现一系列具有支链的烯丙基酯类子的构建。 (Scheme 1c)。
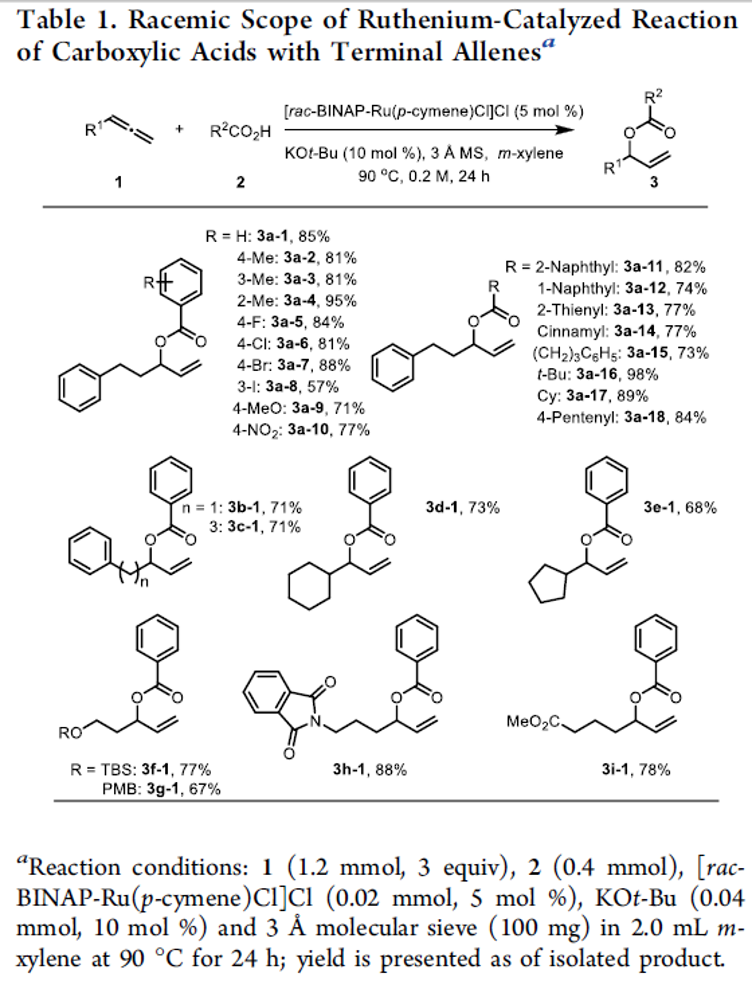
首先,作者采用rac-BINAP-Ru(p-cymene)Cl]Cl作为催化剂,通过反应条件的初步优化,进而成功完成羧酸与末端联烯分子的外消旋烯丙基加成反应过程。同时,该小组进一步对上述烯丙基加成反应方法学中羧酸与末端联烯底物的应用范围进行初步研究 (Table 1)。
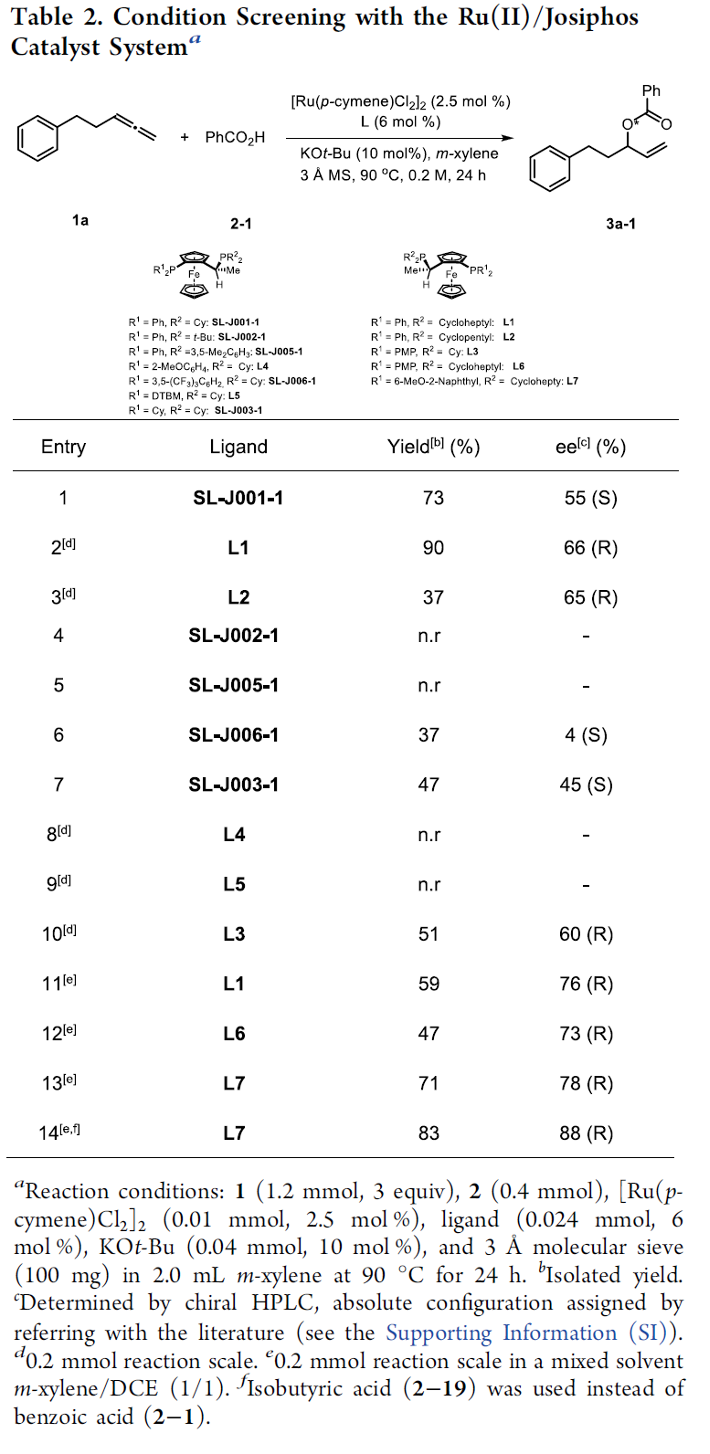
之后,作者开始对钌催化的羧酸与末端联烯之间的不对称烯丙基加成反应策略进行深入研究。首先,作者采用1a与苯甲酸2-1作为模型底物,进行相关反应条件的优化筛选 (Table 2)。进而确定最佳的反应条件为:采用[Ru(pcymene)Cl2]2作为催化剂,L7作为手性配体,KOt-Bu作为碱,间二甲苯/DCE (1:1 v/v)作为反应溶剂,反应温度为90 oC,并以良好的反应收率 (83%)与对映选择性 (88% ee),获得相应的烯丙基加成产物3a-1。
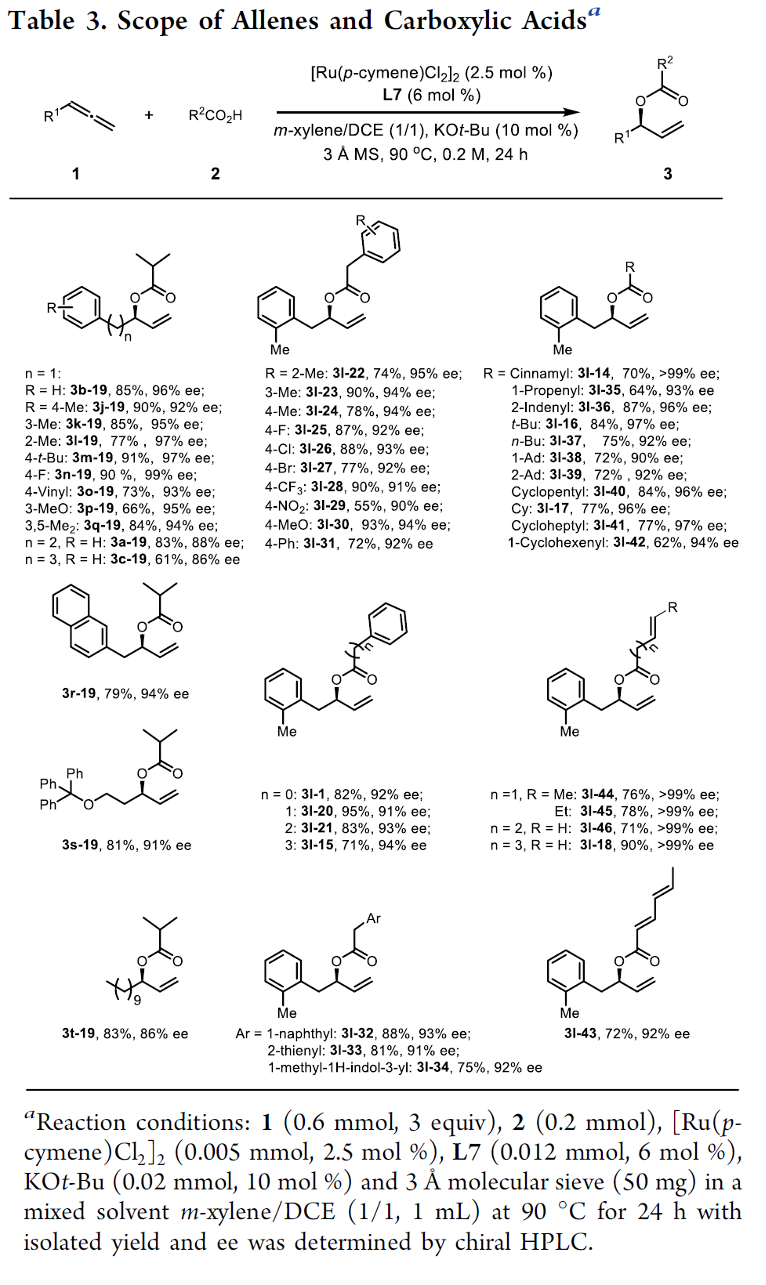
在上述的最佳反应条件下,作者首先对各类联烯底物应用范围进行考察 (Table 3)。研究表明,一系列具有不同链长的联烯底物 (1a–1c),均能够顺利参与上述的不对称烯丙基加成过程,并以优良的对映选择性与良好的反应收率,获得相应的加成产物3a-19–3c-19以及3j-19–3q-19。同时,作者发现,上述的标准反应体系对于邻位、间位以及对位取代的苄基联烯底物 (1j–1q),同样能够以优良的对映选择性与良好的反应收率,获得相应的手性产物3l-22–3l-42。并且,上述的标准反应条件对于1-萘基甲基联烯底物1r,同样能够良好地兼容,并获得相应的手性产物3r–19 (94% ee, 79%收率)。值得注意的是,各类烷氧基 (1s)以及烷基 (1t)基团取代的联烯底物,对于上述的标准反应体系,同样能够表现出良好的兼容性。接下来,作者进一步发现,在选择2-甲基苄基联烯 (1l)作为反应底物时,一系列不同类型的羧酸分子均能够顺利地参与上述的不对称烯丙基加成过程,并以高度的对映选择性控制以及良好至优良的反应收率,获得预期的手性产物。
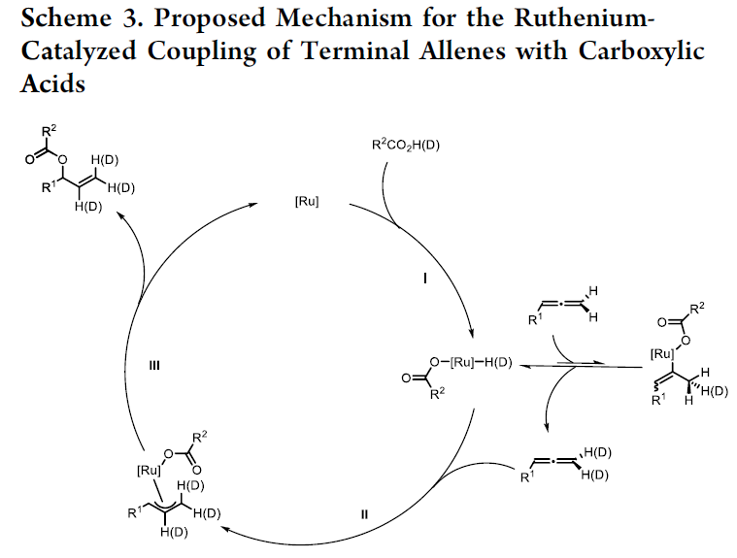
接下来,为提出合理的反应机理,作者进行相关的氘标记实验研究 (Scheme 2)。该小组发现,采用氘代苯甲酸与联烯1l在上述的标准反应体系中进行反应时,能够观察到端烯位置的氘代率为24%,而在2-位的氘代率为23%。同时,研究表明,对于剩余的未参与上述对映选择性加成过程的联烯底物,则在末端位置观察到9%的氘代率。

基于上述实验研究,作者提出一种合理的反应机理 (Scheme 3)。首先,通过羧酸对于手性配合物中Ru(II)中心的氧化加成过程,形成Ru(IV)配合物 (step I)。其中,在末端位置观察到的氘代过程,能够通过在取代基较少的联烯双键位置进行的快速可逆的金属氢化过程以及后续的β-氢消除步骤,而获得合理的解释。之后,通过联烯双键中取代基较多的反应位点进行的金属氢化过程,形成相应的Ru-π-烯丙基配合物 (step II)。再通过后续的还原消除步骤,获得相应的烯丙酯产物 (step III),同时,使Ru(II) 配合物再生,进而完成相关的催化循环过程。
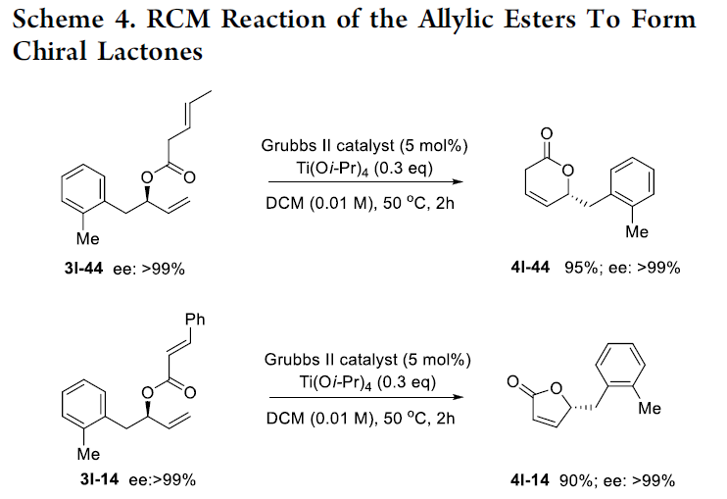
之后,该小组研究发现,相应的烯丙酯产物能够通过后续的RCM反应过程,进一步转化为各类对映纯的内酯化砌块4l-14(90%, >99% ee)以及4l-44(95%, >99% ee) (Scheme 4)。
总结
B. Breit课题组成功设计出一种通过钌催化剂促进的氧化还原中性与原子经济性的羧酸与联烯之间的烯丙基加成反应方法学,进而顺利完成各类支链烯丙基酯衍生物的制备。同时,通过Josiphos配体的精心设计与修饰,进而成功实现对上述烯丙基加成反应过程的区域与对映选择性控制。同时,能够获得良好的反应收率。并且,这一全新的烯丙基加成策略具有广泛的底物范围以及良好的官能团兼容性等优势。
参考文献:
[1] (a) C. A. Sandoval, T. Ohkuma, K. Muñiz, R. Noyori, J. Am. Chem. Soc. 2003, 125, 13490. doi: 10.1021/ja030272c.(b) S. Urban, B. Beiring, N. Ortega, D. Paul, F. Glorius, J. Am. Chem. Soc. 2012, 134, 15241. doi: 10.1021/ja306622y.
(c) A. M. Hayes, D. J. Morris, G. J. Clarkson, M. Wills, J. Am. Chem. Soc. 2005, 127, 7318. doi: 10.1021/ja051486s.
(d) K. Zhang, Y. Yang, H. Liu, Q. Liu, J. Lv, H. Zhou, Adv. Synth. Catal. 2019, 361, 4106. doi: 10.1002/adsc.201900569.
(e) M. Xiang, A. Ghosh, M. J. Krische, J. Am. Chem. Soc. 2021, 143, 2838. doi: 10.1021/jacs.0c12242.
(f) S. I. Kozhushkov, L. Ackermann, Chem. Sci. 2013, 4, 886. doi: 10.1039/C2SC21524A.
(g) T. M. Trnka, R. H. T. Grubbs, Acc. Chem. Res. 2001, 34, 18. doi: 10.1021/ar000114f.
(h) C. Theunissen, M. A. Ashley, T. Rovis, J. Am. Chem. Soc. 2019, 141, 6791. doi: 10.1021/jacs.8b13663.
(i) F. Kakiuchi, P. Le Gendre, A. Yamada, H. Ohtaki, S. Murai, Tetrahedron: Asymmetry 2000, 11, 2647. doi: 10.1016/S0957-4166(00)00244-5.
(j) E. Milczek, N. Boudet, S. Blakey, Angew. Chem. Int. Ed. 2008, 47, 6825. doi: 10.1002/anie.200801445.
(k) Z. Wang, Z. Yin, X. Wu, Org. Lett. 2017, 19, 4680. doi: 10.1021/acs.orglett.7b02320.
(l) A. I. Konovalov, E. O. Gorbacheva, F. M. Miloserdov, V. V. Grushin, Chem. Commun. 2015, 51, 13527. doi: 10.1039/C5CC05436B.
(m) B. P. Machin, J. Howell, J. Mandel, N. Blanchard, W. Tam, Org. Lett. 2009, 11, 2077. doi: 10.1021/ol900454q.
(n) Z. Fan, J. Ni, A. Zhang, J. Am. Chem. Soc. 2016, 138, 8470. doi: 10.1021/jacs.6b03402.
[2] (a) B. M. Trost, M. G. Organ, J. Am. Chem. Soc. 1994, 116, 10320. doi: 10.1021/ja00101a070.(b) N. Kanbayashi, K. Onitsuka, J. Am. Chem. Soc. 2010, 132, 1206. doi: 10.1021/ja908456b.
(c) Y. Suzuki, T. Seki, S. Tanaka, M. Kitamura, J. Am. Chem. Soc. 2015, 137, 9539. doi: 10.1021/jacs.5b05786.
[3] (a) D. J. Covell, M. C. White, Angew. Chem. Int. Ed. 2008, 47, 6448. doi: 10.1002/anie.200802106.(b) E. M. Stang, M. C. White, Nat. Chem. 2009, 1, 547. doi: 10.1038/nchem.351.
(c) K. Takenaka, M. Akita, Y. Tanigaki, S. Takizawa, H. Sasai, Org. Lett. 2011, 13,
- doi: 10.1021/ol201314m.
(b) P. Koschker, A. Lumbroso, B. Breit, J. Am. Chem. Soc. 2011, 133, 20746. doi: 10.1021/ja210149g.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载




















No comments yet.