
本文作者:ChemBoy
导读
在有机合成中,C(sp3)-H键在非金属活化作用下转化成高附加值化合物是一个非常重要的研究领域。最近,德国雷根斯堡大学Burkhard König教授课题组利用商业可得的有机染料2,4,6-三苯基吡啶四氟硼酸盐(TPP)作为光敏剂,在可见光辐射条件下实现了从C(sp3)-H键转化成氰基的方法,通过这种方法可以从甲基(杂)芳烃合成一系列的(杂)芳腈化合物。他们的方法避免了过渡金属催化剂和有毒氰化物的使用,相关研究成果发表在《Angew. Chem. Int. Ed.》上:
“Visible-Light-Promoted Metal-Free Synthesis of (Hetero)aromatic Nitriles from C(sp3)-H bonds”
Kathiravan Murugesan, Karsten Donabauer, and Burkhard König*
Angew. Chem. Int. Ed. 2020, Accepted Articles, 10.1002/anie.202011815 https://doi.org/10.1002/anie.202011815
正文
前言
过渡金属催化剂在一些工业生产、精细化学品和大宗化学品的合成反应中是不可或缺的。在过去二十年里,一些通过热化学或光化学途径进行的C(sp3)-H键活化的过渡金属催化剂已经被发展。其中一种重要的转化就是将石油化工副产品转化成精细化学品,比如从甲苯合成苯腈的转化。在工业上,苯腈是通过甲苯在过渡金属催化(钒)作用下、氨气和氧气高压高温(300-500 ºC)条件下发生胺氧化反应制备。因为氰基不仅是各种药物和生物活性分子中的关键官能团、还是一些精细和大宗化学品的重要合成砌块。所以,近年来,一些新的、条件温和的、过渡金属催化的从醇、胺、醛或是简单CH3合成氰基化合物的反应方法已经被报道[1]。然而,在这些方法中,很多情况下都需要在较高的温度下反应、甚至经常需要使用高毒性的氰化物作为氰基源,或者是需要使用过渡金属催化剂。鉴于这些方法所用试剂的高成本、高毒性等缺点,发展无金属催化、无氰化物作为氰基源、尤其是直接从C-H键转化为氰基的反应方法是非常有必要的。
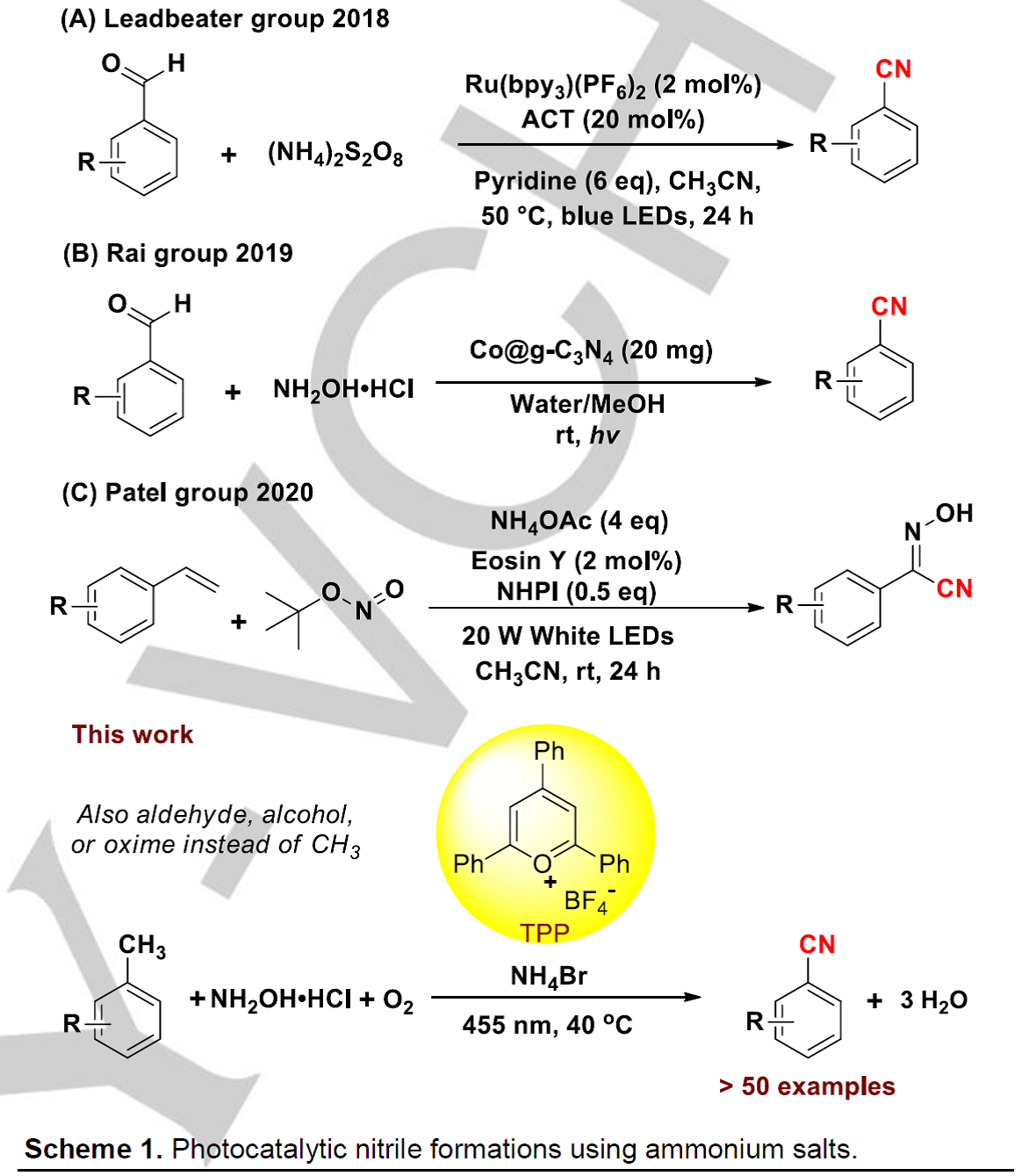
近年来,几例使用更绿色的铵盐作为氮源、通过光化学方法合成腈类化合物的反应得到报道。包括利用过硫酸铵为氮源、芳醛在钌光催化剂作用下生成芳腈(Scheme 1A)[2]或者是芳醛与羟胺盐酸盐在多相光催化剂Co@g-C3N4作用下生成芳腈的反应(Scheme 1B)[3];另外一例非常有意思的反应是苯乙烯转化成氰基肟的反应(Scheme 1C)[4]。这些光催化合成氰基类化合物的反应都成功避免了高毒性氰化物的使用。然而,甲基芳烃直接转化成氰基化合物对于工业的氨氧化反应来说是更简单有效合成路径。
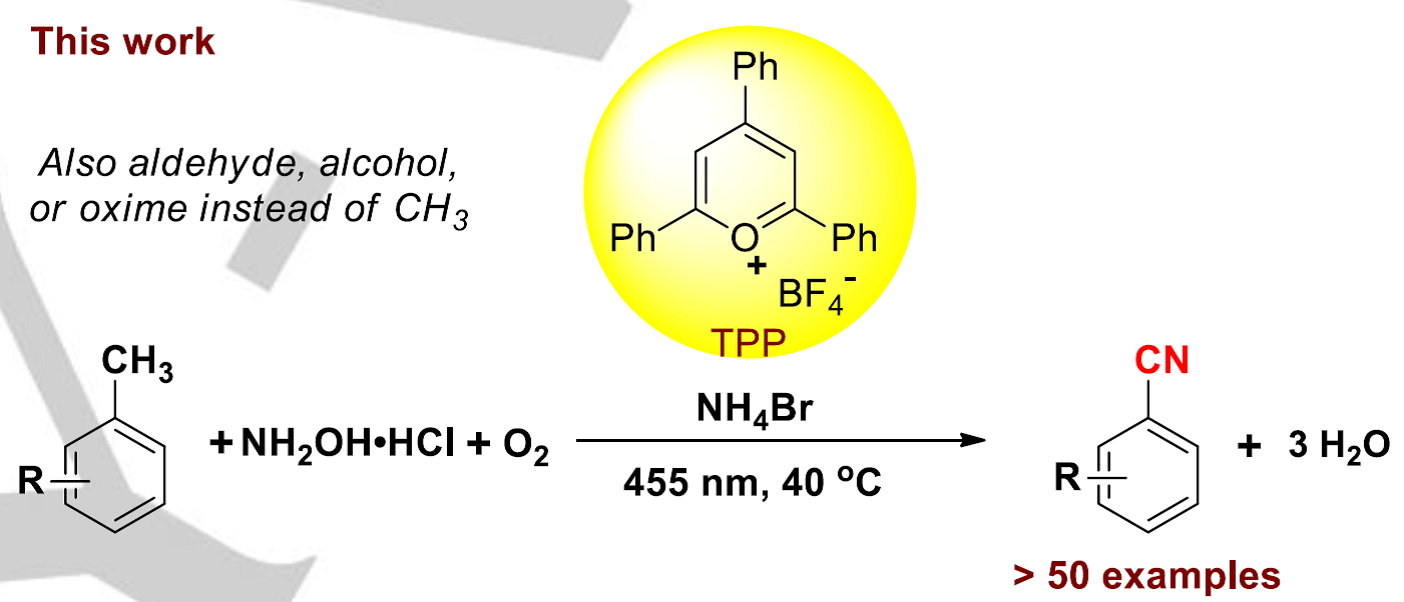
基于以上一些因素,作者设想是否可以利用一种非金属光催化剂、同时无需氰化物作为氰基源,直接将甲苯一步转化为芳腈。作者假设,甲基芳烃(甲苯的氧化电势为E1/2 = +2.36 V vs.SCE)可能会被高氧化性的光催化剂活化或是攫取苄位氢原子。作者在这里利用一种商业可得的有机染料2,4,6-三苯基吡啶四氟硼酸盐(TPP)作为光催化剂、羟胺盐酸盐作为胺源、溴化铵作为添加剂,在氧气氛围中、可见光辐射下实现了从甲苯一步合成芳腈的反应。
条件优化
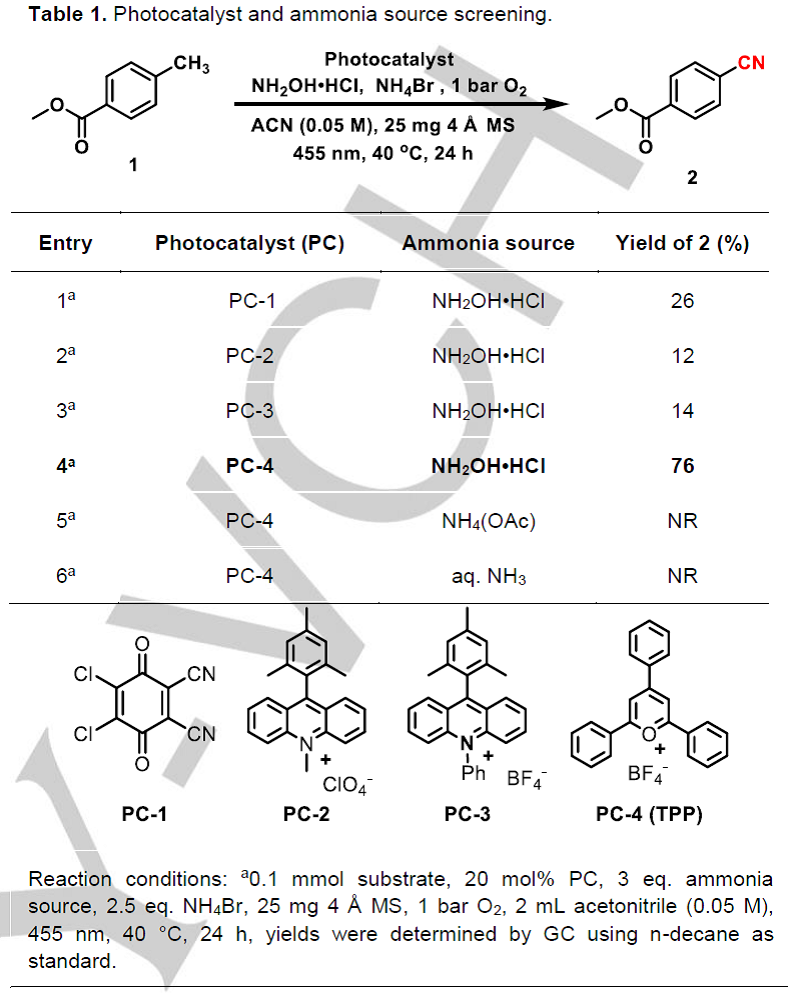
在这里,作者以4-甲基苯甲酸甲酯1作为模板底物对反应条件进行优化(Table 1)。作者以NH2OH•HCl作为氮源、NH4Br作为添加剂、氧气作为氧化剂、乙腈作为反应溶剂,在40 ºC的反应温度、波长为455 nm的可见光照射条件下,对一系列商业可得的有机染料进行了筛选。使用DDQ (PC-1)和吖啶类光催化剂(PC-2、PC-3)时,均能检测到目标产物,但是产率很低(12-26%,Table 1,entries 1-3)。令人高兴的是,当使用TPP(PC-4,E1/2 = +2.55 V vs. SCE)作为光催化剂时能以76%的产率获得目标产物4-氰基苯甲酸甲酯2(Table 1,entry 4)。作者还对其他氮源进行了筛选,如乙酸铵和氨水均没有检测到目标产物(Table 1,entry 5 and entry 6),作者认为这种现象可能是因为这种简单铵离子被氧化的速率比目标底物被氧化的速率快以及PC-4在碱或亲核试剂存在条件下会分解等原因导致的。此外,作者通过控制实验发现,光、光催化剂、氧气和羟胺盐酸盐对该反应的顺利进行至关重要。
在确定了光催化剂后,作者还对反应温度、溴化铵和羟胺的负载量对反应的影响进行了仔细的研究(Fig. 1)。在低温反应条件下,会抑制副产物肟3脱水生成目标产物2(Fig.1a);添加剂溴化铵的存在能够有效促进反应的进行(Fig.1b);此外,无合适的氮源存在条件下,会发生过度氧化反应生成羧酸4(Fig.1c)。值得注意的是,肟3只有在光催化条件下才能转化生成目标产物2(Fig.1d)。
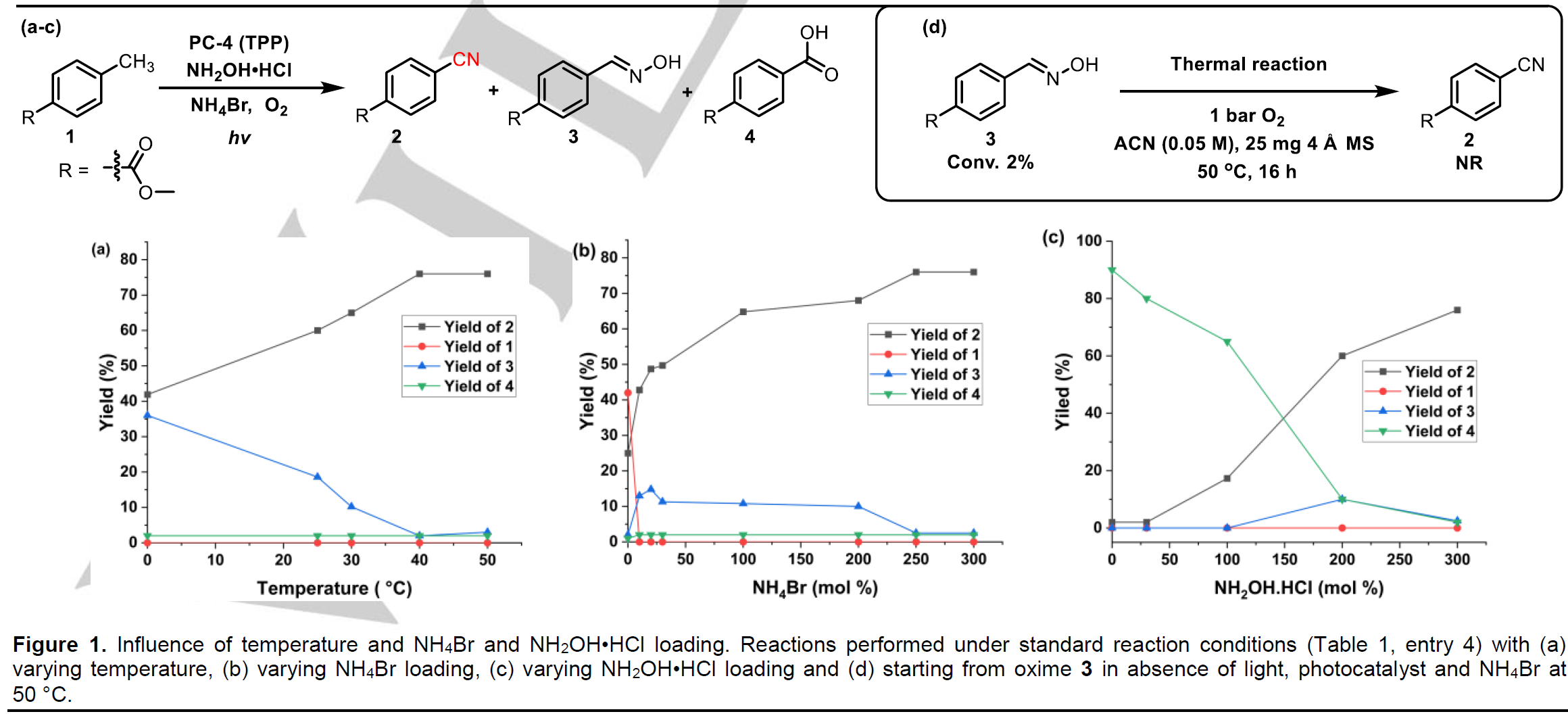
机理假设
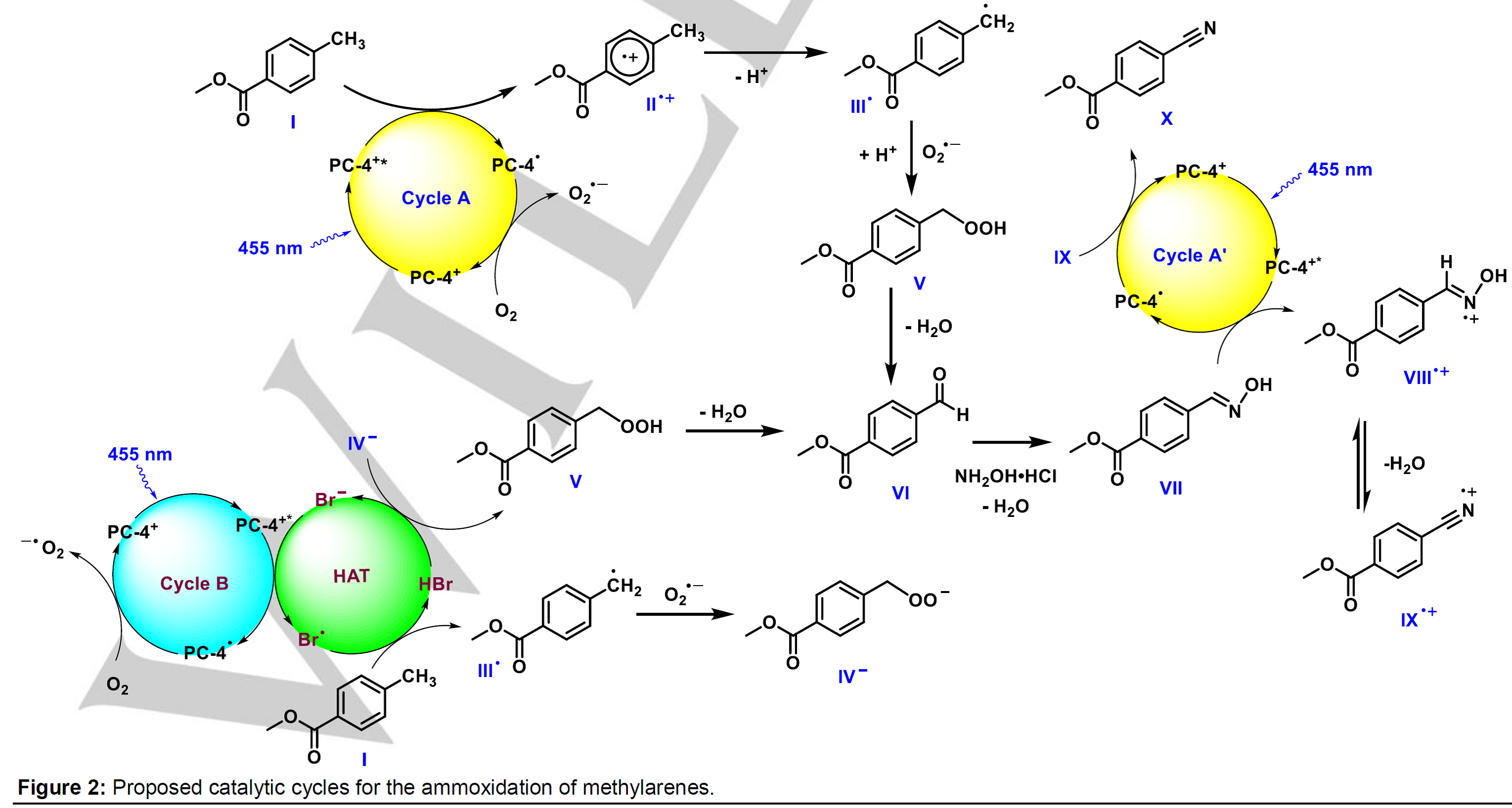
通过条件筛选,作者发现肟3是该反应中的关键中间体;同时,作者发现在没有氧气存在的条件下,该反应不能生成目标产物,这表明反应过程中的肟中间体可能由相应的醛生成。基于这些假设,作者提出了以下可能的催化环(Fig.2, Cycle A):底物1(E1/2(1/1•+) = +2.45 V vs.SCE)被激发态的光催化剂氧化生成自由基阳离子II•+和被还原的光催化剂物种(PC-4•);接着PC-4•与O2发生单电子转移形成超氧负离子O2.-;中间体阳离子自由基II.+失去一个质子生成更稳定的苄基自由基III•,接着与超氧负离子结合同时质子化生成过氧化物V;另外一种可能是苄基自由基III•直接与氧气结合生成过氧自由基,再从PC-4•获得一个电子生成中间体V;中间体V脱水生成醛中间体VI,醛再与羟胺缩合生成肟中间体VII;中间体VII进入光催化循环Cycle A’,VII被激发态的光催化剂氧化生成VIII.+,接着脱水生成IX.+,被还原的光催化剂与IX.+发生单电子转移过程生成目标产物X,同时关闭催化环(Fig.2, Cycle A’)。由于溴化铵在该反应中起到了一种非常关键的作用,所以可能存在另外一种催化循环(Fig.2, Cycle B):在可见光照射下,激发态的PC将Br–负离子氧化成Br•自由基,Br•自由基从底物攫取一个氢原子生成苄基自由基III•和HBr,苄基自由基III.与超氧负离子生成IV-中间体,再被HBr质子化生成中间体V, 中间体V进入Cycle A和Cycle A’所示的循环。作者还通过时间分辨荧光猝灭实验和机理控制实验支撑了以上催化循环假设。
底物拓展
在确定最优反应条件后,作者对甲基芳烃类底物范围进行了探索(Scheme 2)。作者发现无论是吸电子基还是给电子基团取代的底物均能以良好至优秀的产率生成目标产物(Scheme 2, 5-39),如羧酸、酯、酰胺、卤素、氰化物和硼酸酯均能顺利进行反应(Scheme 2, 5-6, 9-12, 15-18 and 22-23);利用这种反应方法可以一锅合成双腈类化合物24;此外,一些具有生物活性和药物活性的复杂分子也能以良好至优秀的产率生成氰基化产物(29-39);有趣的是,一些在氧气存在的光化学反应条件下不稳定的含多个氧原子的底物,如糖类衍生物29与38也能顺利进行反应,分别以62%和60%的产率生成目标产物。而且,一些更复杂的分子,如胆固醇、异冰片、氨基酸以及肽类衍生物也能以45-70%的产率生成相应的目标产物;更有趣的是,消炎药塞来昔布也能以70%的产率生成目标产物(Celecoxib,37);同时,化合物2和塞来昔布(37)在进行克级反应时能分别以65%和60%的产率生成氰基化产物。
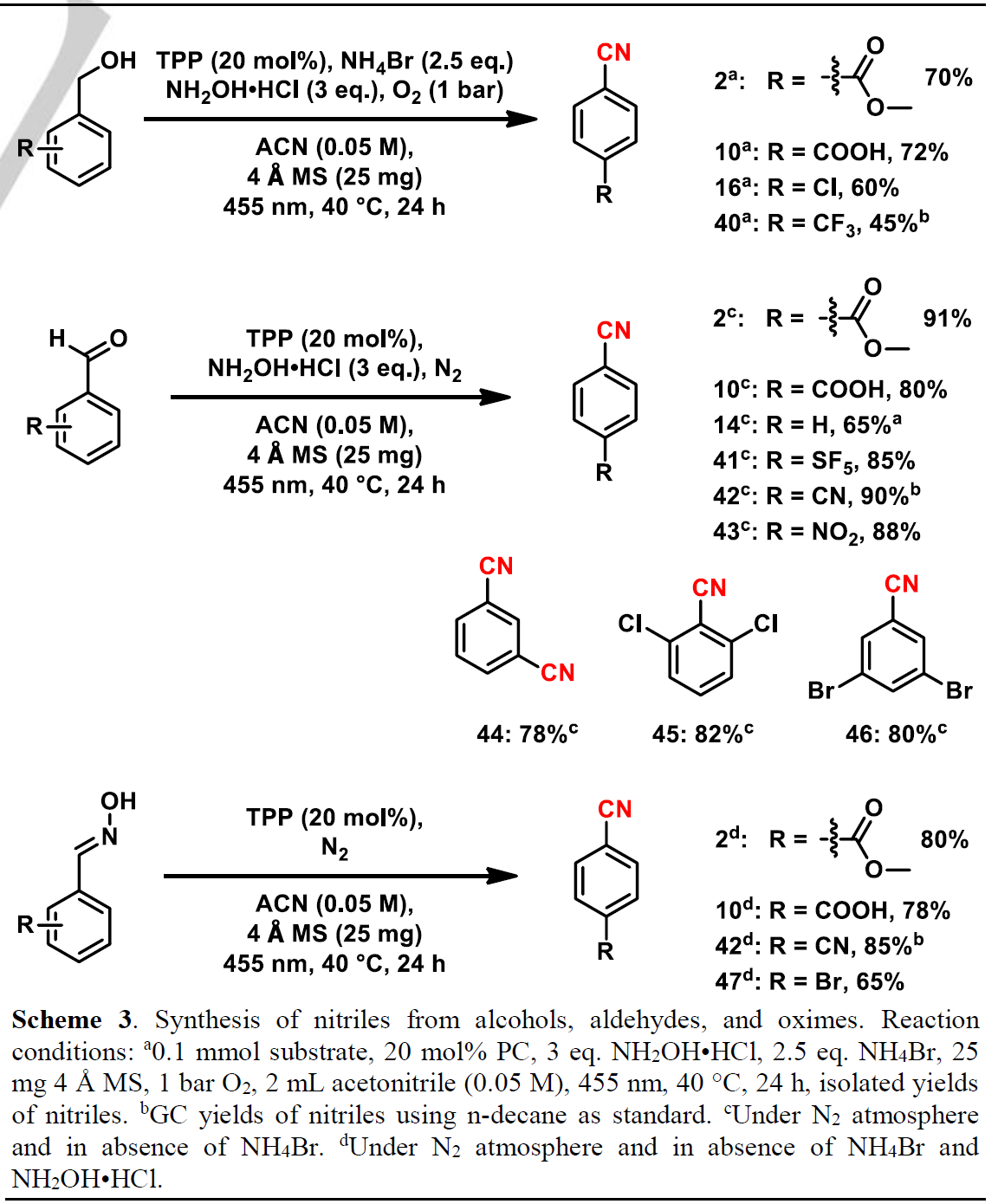
作者在对甲基芳烃类底物进行探索后,继续对该方法在醇、醛和肟类底物的反应适用性进行了探索(Scheme 3)。与甲基芳烃类底物类似,醇、醛和肟类底物在该反应条件下也能以良好至优秀的产率(45-90%)获得相应的氰基化产物。值得注意的是,间苯二甲醛发生两次氰化反应以78%的产率获得间苯二腈产物44;位阻较大的2,6-二氯苯甲醛也能以82%的优秀产率反应获得2,6-二氯苯腈(DCBN, 45)。
总结与评价
Burkhard König教授课题组实现了第一例无金属、氰化物的可见光诱导的有机光催化剂催化的C(sp3)-H键胺氧化反应。该方法以羟胺盐酸盐作为氮源、溴化铵为添加剂、氧气作为氧化剂,在温和的反应条件下就能实现C(sp3)-H键的胺氧化反应,避免了高温、过渡金属和高毒性氰化物的使用;该反应方法的底物普适性较广、官能团兼容性也较好;同时,作者还进行了克级规模反应证明了该反应的实用性。此外,作者还对该反应的机理进行了仔细研究。
参考文献
[1] a) Z. Shu, Y. Ye, Y. Deng, Y. Zhang, J. Wang, Angew. Chem. Int. Ed. 2013, 52, 10573. DOI: 10.1002/anie.201305731; b) J. Liu, H.-X. Zheng, C.-Z. Yao, B.-F. Sun, Y.-B. Kang, J. Am. Chem. Soc. 2016, 138, 3294. DOI: 10.1021/jacs.6b00180; c) W. Zhou, L. Zhang, N. Jiao, Angew. Chem. Int. Ed. 2009, 48, 7094. DOI: 10.1002/anie.200903838.[2] J.Nandi, M. L. Witko, N. E. Leadbeater, Synlett. 2018, 29, 2185. DOI: 10.1055/s-0037-1610272.
[3] F. Verma, P. Shukla, S. R. Bhardiya, M. Singh, A. Rai, V. K. Rai, Catal. Commun. 2019, 119, 76. DOI: 10.1016/j.catcom.2018.10.031.
[4] T. Alam, A. Rakshit, P. Begum, A. Dahiya, B. K. Patel, Org. Lett. 2020, 22, 3728. DOI: 10.1021/acs.orglett.0c01235.














No comments yet.