
本文作者:杉杉
导读
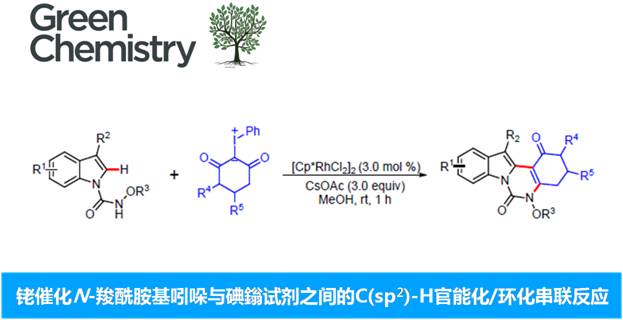
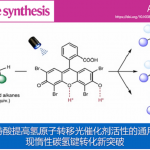
近日,苏州大学纪顺俊与徐小平课题组在Green Chem.中发表论文,报道一种通过铑催化的采用廉价易得的超共价碘鎓试剂 (hypervalent iodonium reagent)与N-羧酰胺基吲哚 (N-carboxamide indole)作为起始原料,进行的C(sp2)-H官能团化/环化串联反应策略,进而成功完成一系列吲哚并喹唑啉酮 (indoloquinazolinone)分子的构建。这一全新的方法学策略具有反应条件温和、优良的反应收率以及良好的官能团兼容性等优势。并且,上述反应在后处理过程中,仅需要通过简单的过滤操作,即可获得较为纯净的目标产物,无需选择实验操作较为繁琐的柱色谱分离操作。值得注意的是,在反应结束后,上述的贵金属催化体系能够有效地进行回收,并能够继续参与相同的催化转化过程 (至少十次),而未表现出相应催化活性的显著降低。进而表明上述反应策略在工业生产中具有良好的应用前景。
Rh(III)-catalyzed C(sp2)-H functionalization/cyclization cascade of N-carboxamide indole and iodonium reagent for access toindoloquinazolinone derivatives
Z. Han, M. Xu, R. Zhang, X. Xu, S. Ji, Green Chem. ASAP. doi: 10.1039/D1GC01820E.
正文
喹唑啉酮 (quinazolinone)以及吲哚骨架单元广泛存在于各类天然产物、药物分子以及具有相关生物活性 (主要涉及抗菌 (antibacterial)、抗癌 (anticancer)以及抗真菌 (antifungal)活性)的有机分子中,例如5-HT3受体拮抗剂 (5-HT3 receptor antagonist)、拓扑异构酶II抑制剂 (topoisomerase II inhibitor)以及荧光核苷 (fluorescent nucleoside) (Scheme 1)。因此,需要开发一种更加高效的反应策略,进而有效地完成一系列吲哚并喹唑啉酮 (indoloquinazolinone)分子的构建。

迄今为止,已经成功设计出诸多构建吲哚并喹唑啉酮类化合物的合成转化策略。然而,却存在需要采用多种贵金属催化剂、反应效率较低、底物难以获得以及后处理过程较为繁琐等诸多弊端[1]。例如,Bao课题组[2]报道通过Cu/Pd接力催化 (relay catalysis)促进的原-偕-二溴乙烯基异氰酸酯 (ortho-gem-dibromovinyl isocyanate)与N-烷基苯胺之间的亲核加成/N-芳基化/C-H活化串联过程,进而实现相应嘧啶并[1,6-a]吲哚-1(2H)-酮 (pyrimido[1,6-a]indol-1(2H)-one)类化合物的构建。Ma课题组[3]报道采用钯催化剂促进的O-氨基苯乙酮与芳基异氰酸酯之间的分子内C-C键形成策略,进而顺利完成一系列吲哚并[1,2-c]-喹唑啉酮分子的构建。Zhang课题组[4]报道通过铑催化的N-羧酰胺基吲哚与氧化锍叶立德 (sulfoxonium ylide)之间的C(sp2)-H活化策略,进而成功完成各类[1,3]oxazino[3,4-a]indol-1-one ([1,3]oxazino[3,4-a]indol-1-one)分子的构建 (Scheme 2)。因此,仍然需要设计一种原料廉价易得,后处理过程较为简洁,催化剂循环率高,并且,环境更加友好的合成转化策略。
碘鎓叶立德 (iodonium ylide)是一种廉价易得的高价碘试剂,目前已经广泛应用于有机合成方法学的实验室研究。其中,在光、热或金属催化剂存在下,碘鎓叶立德能够较为容易地转化为相应的类卡宾 (carbenoid)中间体。同时,带有邻位羰基取代的碘鎓叶立德能够遭受亲核试剂的进攻[5]或通过互变异构化过程,形成相应的亲核反应位点[6]。并且,碘鎓叶立德同样能够应用于原位形成碘自由基,进而引发后续的串联环化过程[7]。同时, 与较为危险易爆的重氮盐化合物相比,碘鎓叶立德化学性质更加稳定,实验操作更加安全。
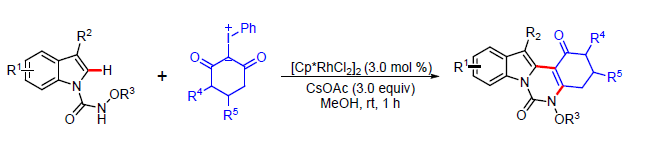
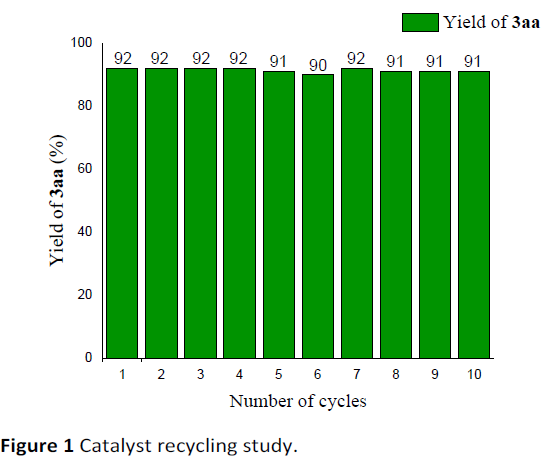
本文中,苏州大学纪顺俊与徐小平课题组研究发现,在[Cp*RhCl2]2与碱存在的条件下,N-甲氧基酰胺吲哚与碘鎓叶立德试剂之间能够通过串联反应过程,进而更加高效便捷地实现相应吲哚并喹唑啉酮分子的构建。上述策略中,无需选择贵金属氧化剂,同时,后处理过程中,仅需要简单的过滤操作,即可获得较为纯净的目标产物。并且,在反应结束后,Rh(III)催化剂能够有效地进行回收,并能够继续参与相同的催化转化过程 (至少十次),同时,未表现出催化活性的显著降低。

首先,作者采用N-甲氧基-1H-吲哚-1-甲酰胺1a与碘鎓叶立德2d作为模型底物,进行相反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用[Cp*RhCl2]2作为催化剂,CsOAc作为碱,在甲醇溶剂中,室温条件下进行反应,最终获得97%收率的产物3aa。

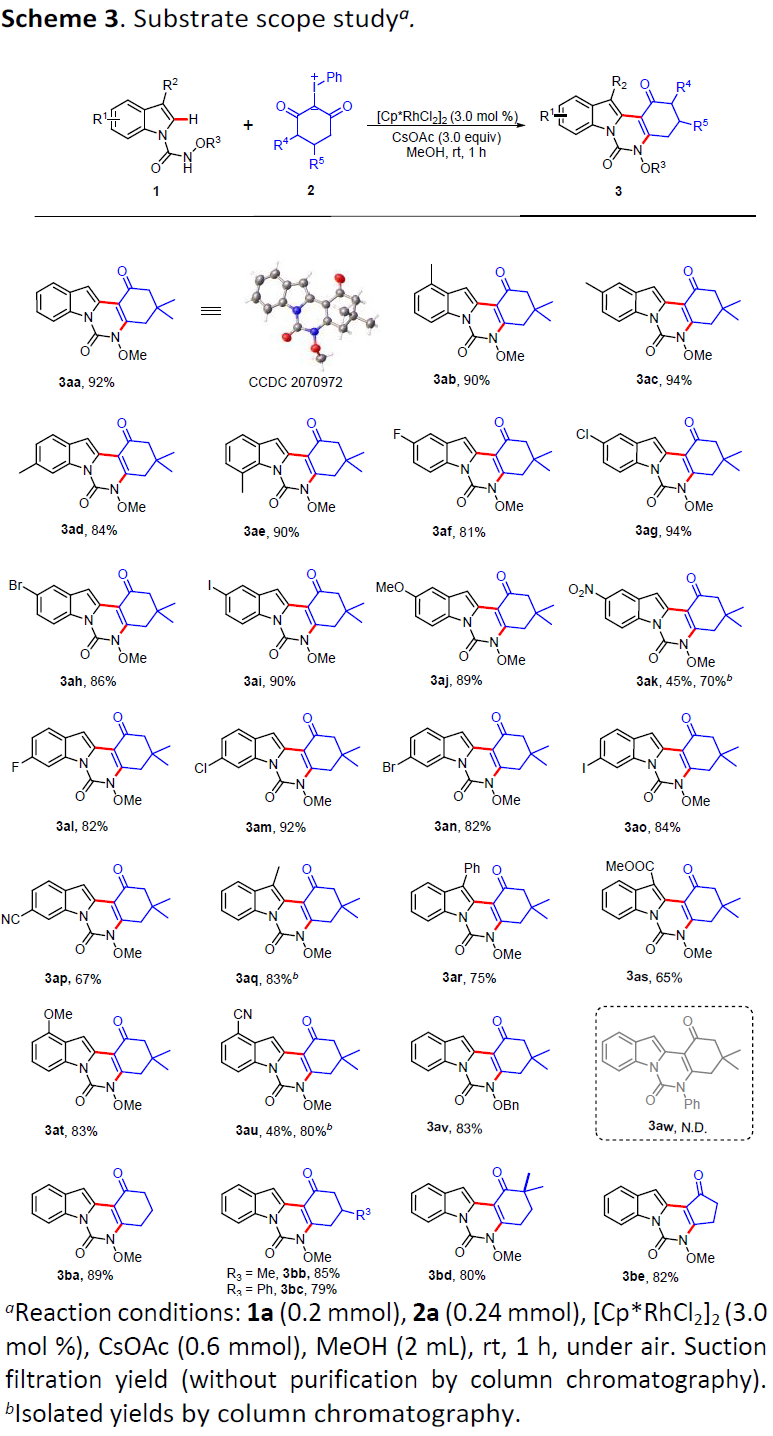
在上述的最佳反应条件下,作者开始对相应吲哚与碘鎓叶立德底物的应用范围进行考察 (Scheme 3)。研究表明,一系列苯环不同位置中具有供电子与吸电子基团取代的吲哚底物,均能较好地与上述的最佳反应条件兼容,并获得相应目标产物3aa–3ap以及3at–3au,收率为45-94%。同时,作者发现,吲哚底物中R2位置的取代基为苯基、甲基以及酯基时,由于立体位阻效应的存在,使目标产物 (3aq–3as)收率略有降低。值得注意的是,苯环中具有4-氰基与5-硝基取代的吲哚底物,经过滤操作之后,却发现目标产物 3ak与3au的收率低于50%,然而,通过柱色谱分离纯化后,目标产物的收率能够分别提高至70%与80%。上述结果表明,产物3ak与3au在甲醇溶剂中具有较好的溶解性。同时,作者观察到,上述的标准条件对于N-苄氧酰胺基吲哚底物同样能够表现出良好的兼容性 (3av),然而,N-苯基酰胺吲哚底物 (3aw),却无法有效地参与上述的串联反应过程。接下来,作者对碘鎓叶立德底物应用范围进行深入研究。实验发现,五种不同类型的碘鎓叶立德底物均能够与上述反应体系良好地兼容,并获得产物3ba–3be,收率为79-89%。
为进一步证实上述反应策略的合成实用性,作者进行相关的克级规模实验 研究 (Scheme 4)。其中,作者观察到,将底物1a的用量扩大至4mmol时,同样能够通过简单的过滤操作,获得良好收率 (90%)的目标产物3aa。由此表明,上述的串联反应策略,能够有效地应用于工业生产过程。
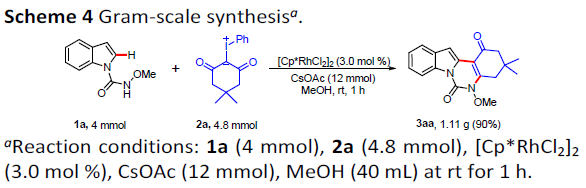
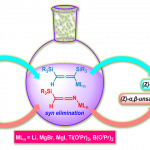
接下来,作者在上述标准反应条件下,进一步考察铑催化剂在甲醇溶剂中的再循环性 (Figure 1)。研究表明,在过滤除去产物之后,有机相中的催化体系能够有效地回收,并能够重新参与相同的催化反应过程。同时,在重复参与相同的催化反应过程十次之后,并未观察到催化活性的显著降低。
总结
苏州大学纪顺俊与徐小平课题组报道一种铑催化的C(sp2)-H官能团化/环化串联反应方法学,能够在较为温和的反应条件下,通过高价碘鎓试剂与N-羧酰胺基吲哚作为起始原料,成功完成一系列吲哚并喹唑啉酮衍生物的构建。同时,这一全新的反应策略具有较高的目标产物收率、良好的官能团兼容性、后处理过程简洁等优势。值得注意的是,在反应完成后,相应的贵金属催化剂能够有效地进行回收,并能够继续参与相同的催化转化过程 (至少十次)。 进而表明上述策略在工业生产过程中具有良好的应用价值。
参考文献
[1] (a) J. Liu, M. Shen, Y. Zhang, G. Li, A. Khodabocus, S. Rodriguez, B. Qu, V. Farina, H. Senanayake, B. Lu, Org. Lett.2006, 8, 3573. doi: 10.1021/ol061440j.(b) I. Nukamura, Y. Sato, M. Terada, J. Am. Chem. Soc. 2009, 131, 4198. doi: 10.1021/ja900174t.
[2] Z. Wang, J. Yang, F. Yang, W. Bao. Org. Lett. 2010, 12, 3034. doi: 10.1021/ol101041e. [3] Y. Ding, H. Yan, R. Chen, X. Xiao, Z. Wang, L. Wangand, Y. Ma, J. Org. Chem. 2021, 86, 1448. doi: 10.1021/acs.joc.0c02155. [4] H. Xie, M. Zhong, H. Kang, B. Shu, S. Zhang. Adv. Synth.Catal. 2021, 363, 1436. doi: 10.1002/adsc.202001380 [5] (a) S. Mayakrishnan, M. Tamizmani, N. U. Maheswari, Chem.Commun. 2020, 56, 15462.doi:10.1039/D0CC06038K.(b) S. Nunewar, S. Kumar, H. Pandhare, S. Nanduri, V. Kanchupalli, Org. Lett. 2021, 23,4233.doi:10.1021/acs.orglett.1c01167.
[6] (a) X. Huang, Y. Liu, Y. Liang, S. Pi, F. Wang, J. Li. Org. Lett. 2008, 10, 1525. doi: 10.1021/ol800051k.(b) R. M. Moriarty, S. Tyagi, D. Ivanov,and M. Constantinesc, J. Am. Chem. Soc. 2008, 130, 7564. doi: 10.1021/ja802735f.
[7] J. Tao, C. D. Estrada, G. K. Murphy, Chem. Commun. 2017, 53, 9004. doi: 10.1039/C7CC04859A.














No comments yet.