
本文作者:杉杉
导读
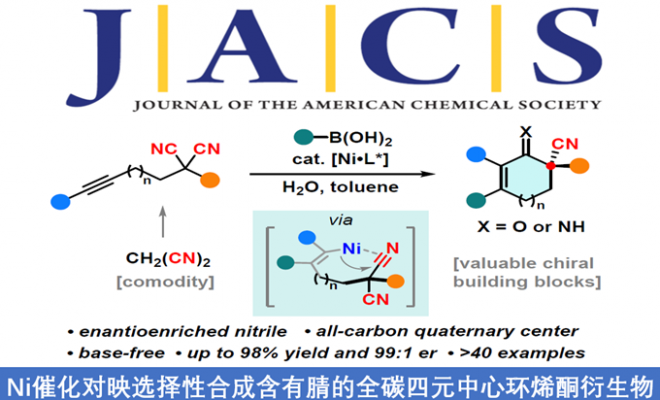
手性腈作为现代有机合成和药物中重要的结构,通过对丙二腈衍生物中两个腈基的选择性区分作为合成手性腈最为直接的方法,然而相对的研究却很少。近日,武汉大学刘文博教授课题组在美国化学学会杂志(Journal of the American Chemical Society)发表论文,通过镍催化,可实现丙二腈衍生物的对映选择性去对称化反应,从而获得含有腈的全碳四元立体中心化合物。该方法涉及芳基-镍络合物与炔烃的加成,然后腈进行选择性的插入,从而获得多种五-七元环的手性腈基环烯酮衍生物。此外,通过克级实验和产物的后期修饰,进一步证明了该反应的实用性。
Enantioselective Assembly of Cycloenones with a Nitrile-ContainingAll-Carbon Quaternary Center from Malononitriles Enabled by Ni-Catalysis
Zhiwu Lu, Xu-Dong Hu, Hui Zhang, Xiao-Wen Zhang, JinhuiCai, Muhammad Usman, HengjiangCong, and Wen-Bo Liu
J. Am. Chem. Soc. ASAP DOI: 10.1021/jacs.0c02075
正文
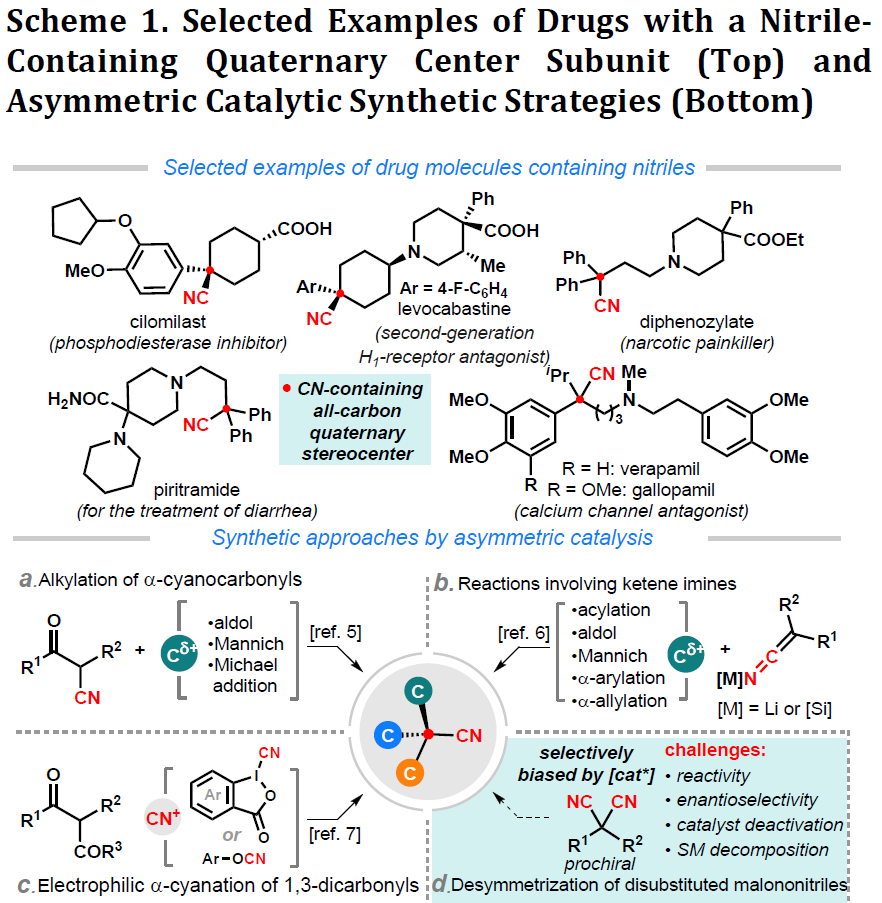
含有腈基团的化合物,在药物中具有重要的作用。生物相容性以及代谢稳定的腈基团,具有促进极性相互作用、增强氢键键合性质、改善分子的毒理学特征等优点。据文献报道,含腈基团的药物已广泛存在于各类临床候选药物中。而具有生物活性的腈基化合物特别重要(Scheme 1),可防止腈在α-碳上的氧化,避免有毒氰化物的释放。此外,腈也是有机合成中最有用的官能团之一,可以轻松转化为一些通用的官能团,例如羧酸、醛、胺、恶唑啉和哌啶等。因此,一些文献也报道了相关的催化方案,用于合成含腈的全碳四元立体中心化合物,如α-氰基羰基的亲电官能化(Scheme 1a)、乙烯酮亚胺的亲电官能化(Scheme 1b)、1,3-二羰基的α-氰化反应(Scheme 1c)等。
去对称化反应作为合成全碳四元中心的有效工具,而丙二腈(两个腈)的不对称化作为构建手性腈最为直接的方法,但报道却很少。可能是由于其对过渡金属的配位亲和力和腈基的微小空间的差异,从而导致丙二腈的立体选择性具有一定的挑战。同时,使用过渡金属催化剂或有机金属试剂时,容易导致丙二腈发生脱氰分解的弊端。尽管丙二腈的对映选择性酶水解反应已有数十年的历史,但迄今为止,仅揭示了丙二腈化学催化去对称反应的两个例子,即铑催化的[2+2+2]环加成反应(Scheme 1d)和钌催化的水合反应。然而,这些文献存在底物范围窄、对映选择性较差、使用贵重过渡金属催化剂等问题。
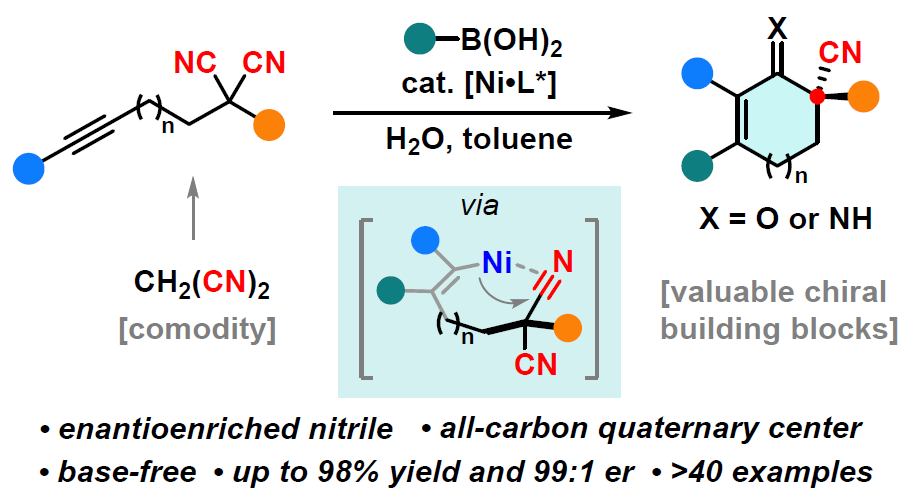
为了解决此类问题,同时受Lam课题组等报道的关于过渡金属催化的炔烃插入/加成反应的启发(Scheme 1e),作者设想了可经分子间炔烃的加成和分子内环化串联环化过程,从而促进丙二腈的去对称化,获得手性腈的产物(Scheme 1f)。在本文中,作者报道了在镍催化条件下(无碱),可使炔基固定的丙二腈衍生物1与芳基硼酸2进行去对称化反应,从而获得高对映选择性和产率的含有腈的全碳四元中心产物3。

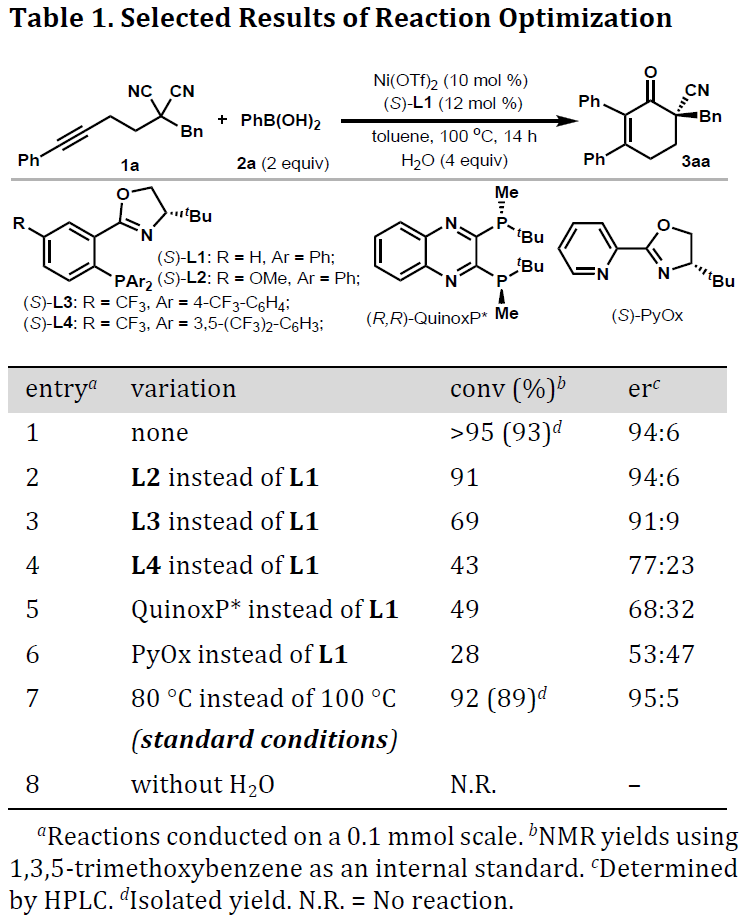
首先,作者以炔基丙二腈化合物1a和苯硼酸2a作为模型底物,进行了相关去对称化反应条件的筛选(Table 1)。当使用Ni(OTf)2/膦基恶唑啉(phox,L1)作为催化剂时,获得94:6 er和93%收率的产物3aa(entry 1)。同时,使用富电子的膦配体L2时,也获得较好的结果(entry 2),但使用缺电子的膦配体L3和L4时,反应性和对映选择性大幅下降(entries 3-4)。当使用双膦或二氮配体时,收率和er均较差(entries 5-6)。最后,将反应的温度降至80℃时,可获得89%的产物3aa,对映选择性也略有提高(95:5 er),作为此反应的最佳条件(entry 7)。值得注意的是,水对于该反应至关重要,当反应中不存在水时,则不发生反应(entry 8)。
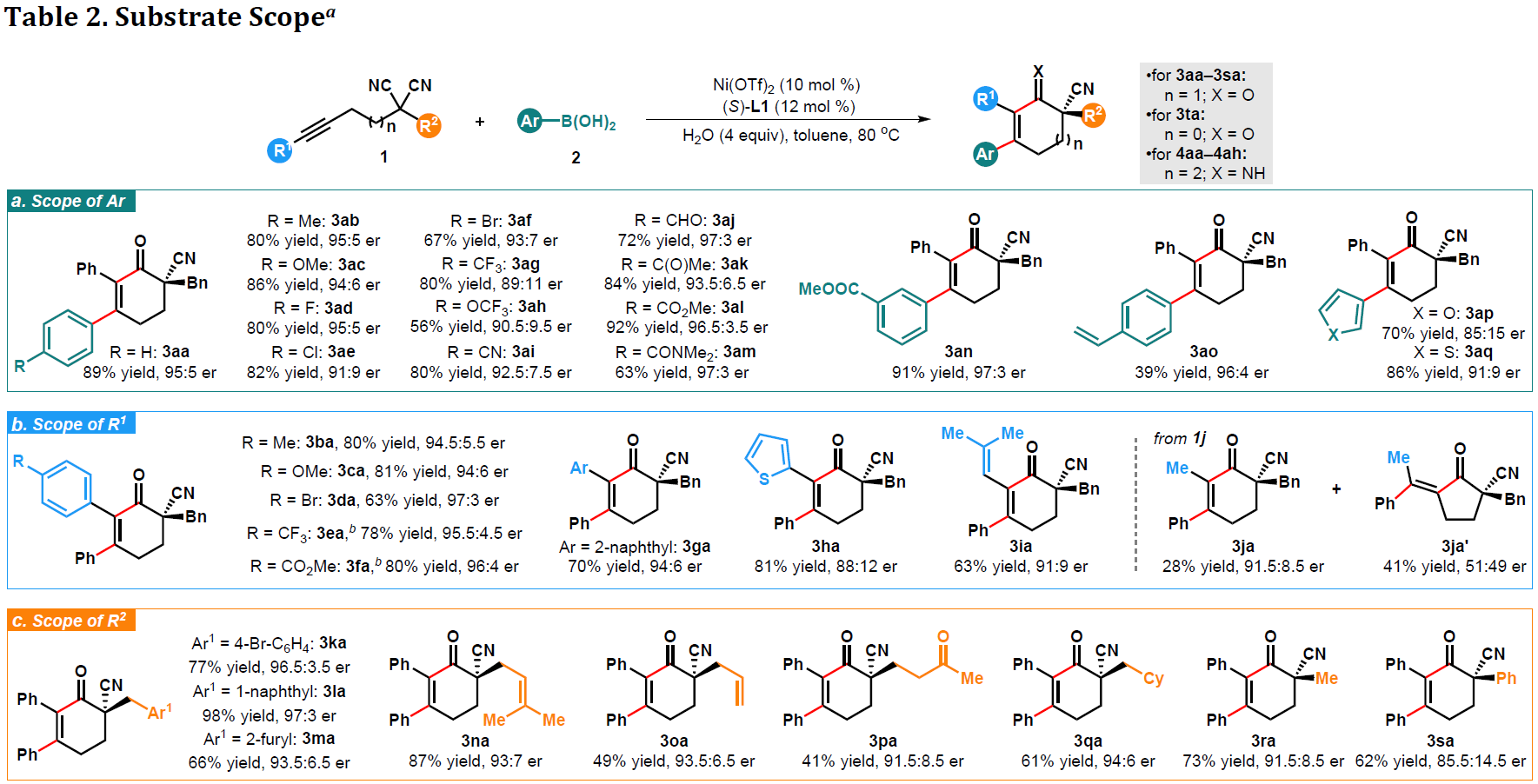
在获得上述最佳反应条件后,作者开始对底物进行了扩展(Table 2)。首先,作者对芳基硼酸的底物范围进行了相关的研究(Table 2a)。当带有给电子基团(如甲基、甲氧基)时,获得相应产物3ab和3ac。同样带有卤素基团时,也可获得相应的产物3ad–3af。值得注意的是,一些常用的官能团,如三氟甲基、三氟甲氧基、氰基、醛、乙酰基、酯、酰胺等,均可获得相应的产物3ag-3am。此外,间位取代的底物2n也可获得91%收率和97:3 er的产物3an。然而,使用末端带有烯烃的苯基硼酸时,产物3ao收率有所降低(er为96:4)。而使用杂芳基亲核试剂(如3-呋喃基和3-噻吩基硼酸),产物3ap和3aq的对映选择性降低,但收率较好。
随后,作者对炔烃丙二腈底物中R1进行了扩展(Table 2b)。反应结果表明,该反应不受电子效应的影响,各类给电子基团(-Me、-OMe),吸电子基团(-Br、-CF3、-CO2Me),均可进行反应,获得63-81%收率的产物3ba–3fa(er为94:6-97:3)。此外,使用具有缺电子的芳基取代炔烃底物时,在80℃时反应缓慢,但升高温度至100℃时,产率可提高。当使用萘炔烃底物时,也能够获得70%收率的产物3ga,但er 为70:6,同样杂芳基(噻吩基)和烯基取代的炔烃也与体系兼容,获得63-81%收率的3ha和3ia,但er减少了。而将取代基R1改为甲基时,得到异构体3ja和3ja’的混合物。
紧接着,作者又对丙二腈中α-位置的R2取代进行了研究(Table 2c)。使用4-溴苄基、1-萘甲基和2-呋喃基甲基时,可获得66-98%收率的3ka-3ma(er为93.5:6.5-97:3)。通过X-射线分析进一步确认了确定3ka(er > 99:1)的绝对构型。同时,使用含有烯丙基和3-氧代丁基的底物时,也可获得相应的产物3na–3pa,为进一步修饰提供了多种可能。此外,脂肪族α-取代(环己基甲基、甲基)的丙二腈产物3qa和3ra也被获得。
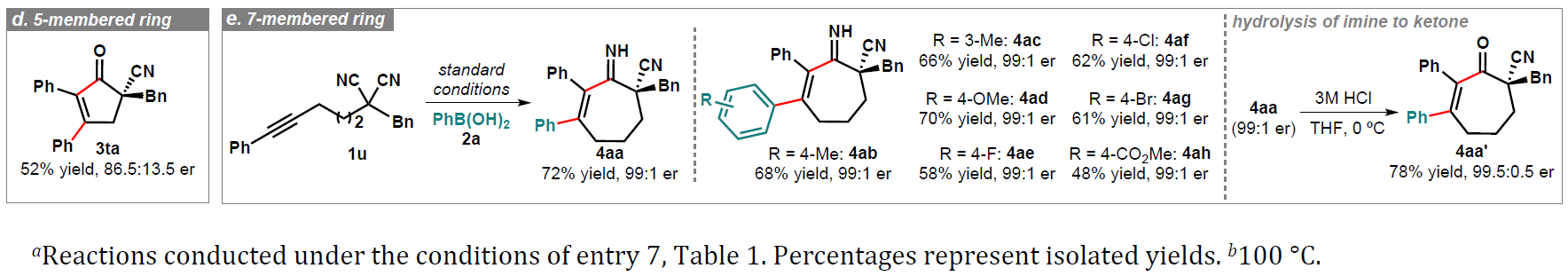
随后,作者还进行了相关的环扩张研究(Table 2d-2e)。当使用1t作为底物时,生成环戊烯酮3ta,产率为52%,er为86.5:13.5。有趣的是,七元亚胺4能够以足够的收率形成具有高对映选择性(99:1 er)的色谱分离稳定剂。在标准条件下,丙二腈1u和苯基硼酸2a反应,可以获得 72%的收率和99:1er的环庚-2-烯-1-亚胺产物4aa。此外,给电子基团(-Me、-OMe)和吸电子基团(-F、-Cl、-Br、-CO2Me)取代的芳基硼酸,均与体系兼容,从而获得具有出色对映选择性(er 为99:1)的产物4ab–4ag。而在0℃下用3M HCl酸解后,亚胺很容易水解为相应的酮4aa’,而不会损失er。
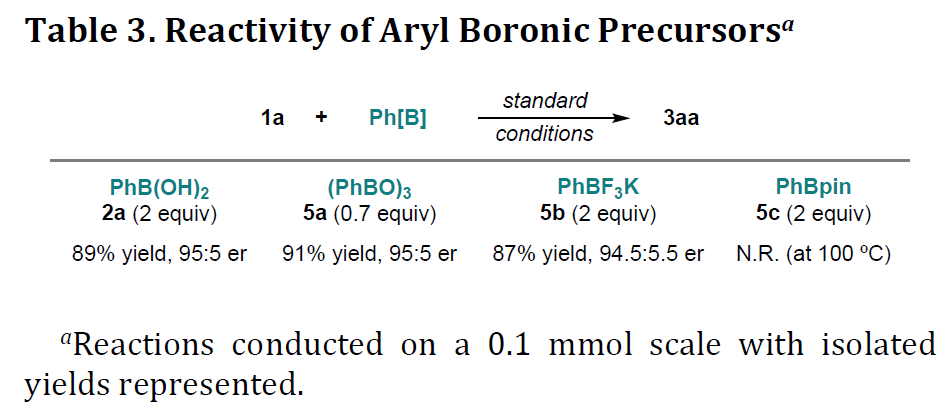
此外,作者还使用丙二腈衍生物1a与其他芳基硼源进行相关的验证(Table 3)。除苯硼酸2a外,1a还可与三苯基硼氧烷5a和三氟硼酸苯酯5b反应,获得高收率和对映选择性的产物3aa。但是,使用PhBpin(5c)时,未能获得所需的产物,可能是由于在无碱条件下缓慢的金属转移作用所致。
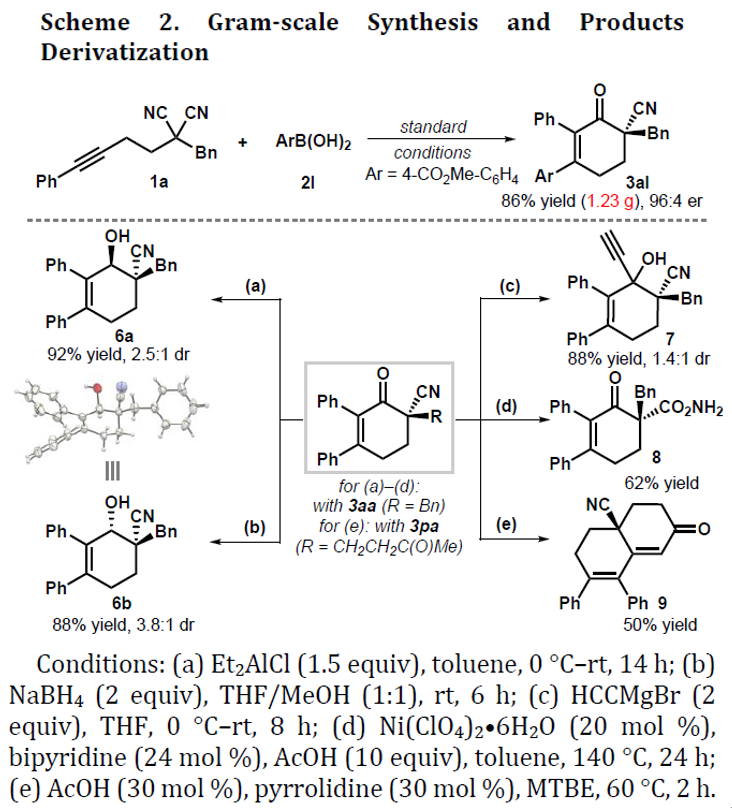
紧接着,作者对反应的实用性进行了相关的研究(Scheme 2)。首先,1a的克级实验,同样获得86%收率和96:4 er的产物3aa。而通过选择性还原剂可以实现反式(6a)和顺式(6b)醇的合成。同时,若将含乙炔基的格氏试剂与3aa中的酮进行加成时,可获得具有两个连续立体中心的产物7。此外,3aa中的腈基可经水解获得酰胺8,产率为62%。若使用带有甲基酮的产物3pa,可经醛醇缩合获得中等收率的带有γ-季中心双环产物9。
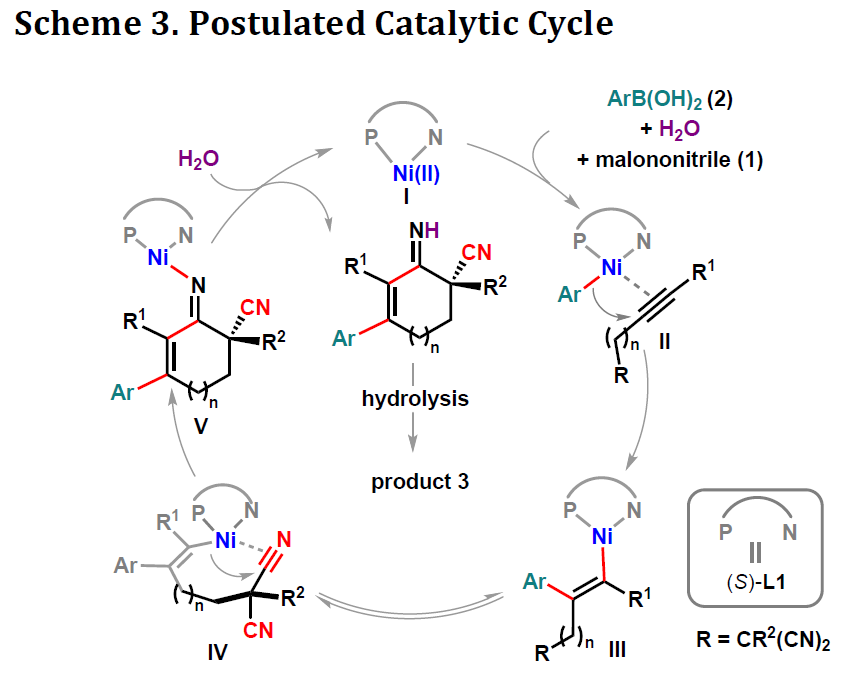
根据相关的对照实验以及文献的查阅,作者提出了一种可能的反应机理(Scheme 3)。首先,在底物1存在下,镍配合物I与芳基硼酸2经重金属化生成芳基-镍络合物II。紧接着,炔烃插入至C-Ni键中生成顺式烯基-镍络合物III,顺式/反式异构化的可逆性从而获得反式烯基-镍络合物IV。随后,其中一个腈基经加成生成亚氨基-镍络合物V,作为催化循环中对映异构的步骤 。最后,V经质子化后产生活性镍催化剂和亚胺,亚胺经水解生成环烯酮产物3。
总结
武汉大学刘文博教授课题组报道了,一种镍催化炔烃的加成和串联环化反应,具有区域选择性和对映选择性,并合成多种含腈的全碳四元中心的环烯酮衍生物。此外,通过市售的Ni(OTf)2和phox配体L1作为催化剂和配体,实现丙二腈衍生物中两个腈基的去对称化反应,获得中等以上收率和高对映选择性的相应的产物。同时,通过克级实验以及产物的后期修饰,进一步证明了该反应的实用性。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
















No comments yet.