氢化硅烷、特别是三甲基硅烷、四氢硅烷是有特殊臭味的无色有毒气体。由于这两种物质很容易自燃而且易爆,所以在使用时候常常会引起各种事故[1],在取用的时候尤其需要注意。通常情况下,对于没有条件配备防护措施的实验室来说,做这类实验就很困难了。
很多时候就因为这样一些困难,使得反应做不了,进而导致可能很有意义的发现无法实现,这确实是十分可惜的事情。
难道就真的没有能够很简单安全合成氢化硅烷的方法么?
这次就让我们一起来看一下最近柏林工业大学Oestreich等人的研究「简单合成不稳定的氢化硅烷的方法」。首先先向大家简单介绍一下氢化硅烷在有机合成化学中的用途。
氢化硅烷的有用性:硅氢化反応
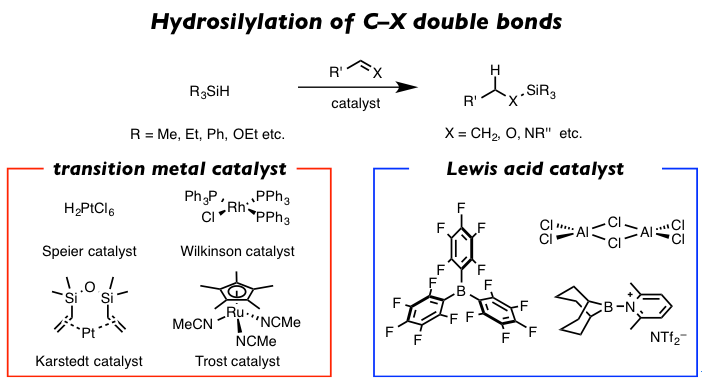
图1 硅氢化反应及所需催化剂
首先最常用的就是一些硅氢化反应的应用。硅氢化反应顾名思义就是在有机骨架上导入硅烷基的最基本的手法之一(图 1)。对于如烯烃、酮、烯胺等拥有不饱和双键的底物,在过渡金属催化剂或者路易斯酸的催化下,会发生硅氢化加成反应。
该类反应以1956年的Pt催化剂(Speier催化剂)催化的烯烃的硅氢化反应为开端、进而发展到Pd[2]、Rh[3]、Ru[4]等多种过渡金属催化剂、以及氯化铝[5]、三(五氟苯基)硼烷(B(C6F5)3) [6]等路易斯酸催化的各种类似反应被相继报道。但是上述中作为反应试剂的氢化硅烷都具高毒性与易燃性,特别是低分子量的四氢化硅,在常温下是气体,所以在取用上是反应最大的难点。
那么我们就来看看Oestreich等人是用怎样的方法来合成并纯化这种不稳定的硅烷类化合物的。
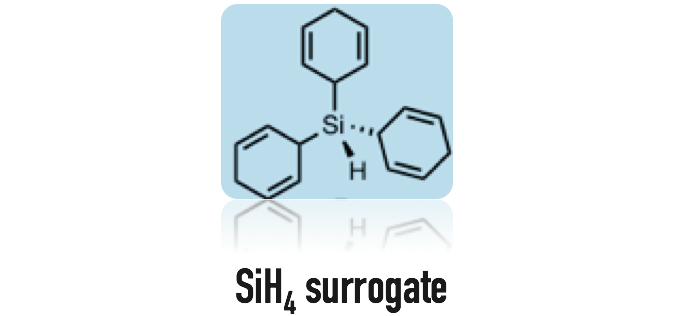
稳定的容易取用的甲基硅烷的前体1的开发
实际上,该方法不是直接合成低分子量的氢化硅烷,而是开发可以再在反应中简单生成目标氢化硅烷的前体。在2013年,这个设想被成功实现,液体状的取用简单的Me3SiH的前体1被成功开发并应用(图 2)[7]。这个前体的设计最早是从B(C6F5)3催化的氢化硅烷反应中得到的启发。
在论文报道中,我们可以看到下图中,底物1在催化量的B(C6F5)3存在下能与烯烃发生氢化硅烷化反应,事实上烯烃是与反应体系中生成的Me3SiH发生的反应。在这之后,在B(C6F5)3催化下,1与酮或者烯胺的同类反应也被相继报道[9]。
在机理方面,经过研究发现,在普通的B(C6F5)3催化的使用Me3SiH的氢化硅烷化反应中(下图右),首先高位阻的路易斯酸B(C6F5)3能够与硅烷形成B-H结合,通过这个相互作用,路易斯酸形成复合体2(进程I)活化Si-H键[8]。然后复合体2的硅烷基与不饱和键进行加成反应(进程II),生成的碳正离子再通过捕捉氢后完成氢化硅烷化反应(进程III)。Oestreich等人就是以这个反应作为基础,把Me3SiH的氢原子替换成「1,4环己二烯基」形成Me3SiH的前体1,尝试完成这个反应。
详细的说就是(下图左),1,4环己二烯基的4位的氢原子与路易斯酸的硼原子进行配位活化(进程IV)形成silylarenium负离子3(进程V)。接着,由于3的芳香化作用形成与苯环配位的silylium离子。然后4再与硼上的氢加成生成Me3SiH的同时也再次形成催化剂B(C6F5)3。(进程VI)
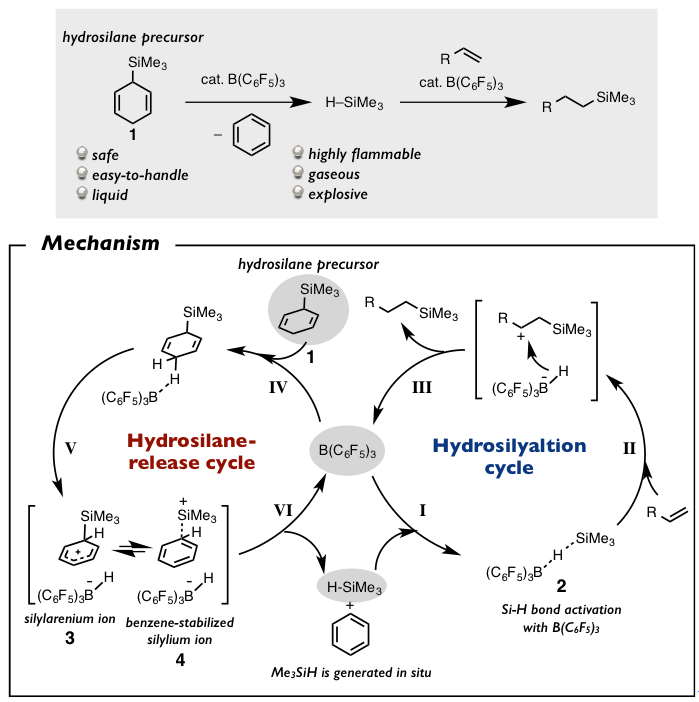
图2 Me3SiH前体1与反应机理解析
稳定的四氢化硅烷前体5、6的开发及在氢化硅烷化反应上的应用
进而,研究人员着手开发在体系中生成四氢化硅烷(SiH4)的方法。也是在最近,SiH4前体5、6被成功合成,并且被应用于氢化硅烷化反应中[10]。
作者首先参考Me3SiH的前体1,把SiH4的两个或者氢原子替换成1,4-环己二烯基,分别形成SiH4前体5和6。对于5或6的合成方法,如下图所示,2,5-环己二烯锂与HSiCl3或者(EtO)2SiCl2进行亲核取代经过一步或者两步制备获得,并且这两个物质可以通过重结晶纯化,十分稳定。合成得到的5与6在催化量的B(C6F5)3作用下在体系中分别能够生成7或SiH4,这两个中间体通过NMR测定被验证。从而这两个前体5、6也被尝试应用于氢化硅烷化反应中。
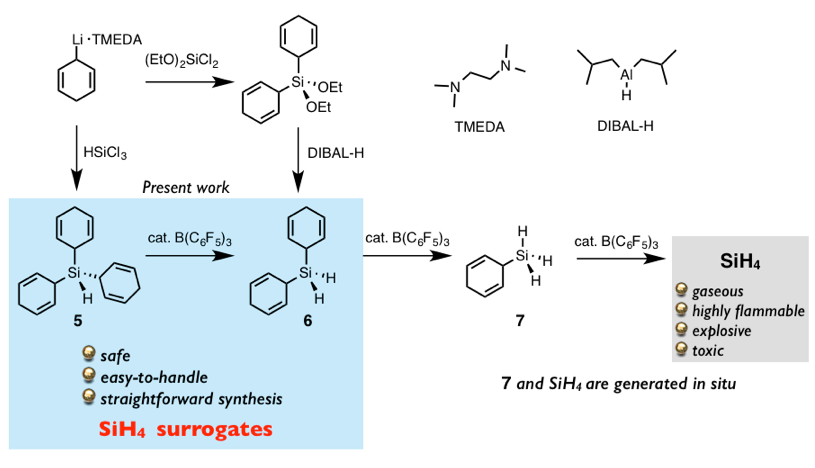
接下来,作者利用前体5与各种烯烃进行氢化硅烷化反应(图3a)。在与苯乙烯,环状、链状的烯烃的反应中发现,根据底物的立体位阻不同,发生氢化硅烷化的次数也不同,一般是2-4次的产物为主。另外在与3-己炔的氢化硅烷化反应得到了反式加成产物。另外5与铂金催化剂首先进行1-辛烯的氢化硅烷化,然后再在B(C6F5)3下进行1,4-环己二烯基的脱保护,最终合成了单取代的氢化硅烷8(图 3b)。
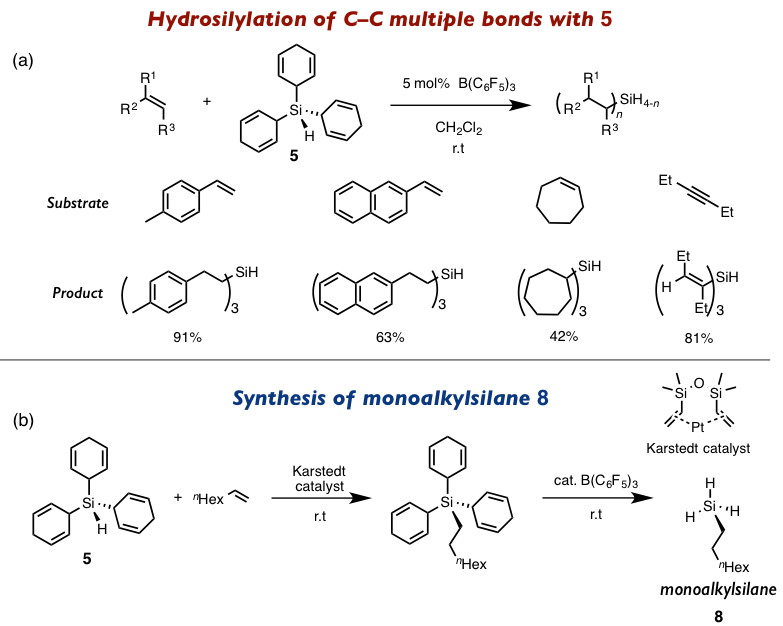
图3 氢化硅烷前体的开发以及应用
综上,这回介绍的是低分子氢化硅烷类前体的简便合成法以及利用在体系中形成的SiH4再继而进行的烯烃或炔烃的氢化硅烷化反应的应用。本论文最大的亮点就是使得本来具有危险性取用很困难的SiH4类化合物可以很安全的制备获得并且应用于反应中,是十分具有实用性的一个发现。小编在这里也希望借助这一发现能够使得硅化学有更大的发展。
参考文献
- (a) Chen, R. J.; Tsai, Y. H.; Chen, K. S.; Pan, R. H.; Hu, C. S.; Shen, C. C.; Kuan, M. C.; Lee, C. Y.; Wu, C. C. Process Saf. Prog. 2006, 25, 237. DOI: 10.1002/prs.10136 (b) Chang, Y. Y.; Peng, J. D.; Wu, C. H.; Tsaur, C. C.; Shen, C. C.; Tsai, Y. H.; Chen, R. J. Process Saf. Prog. 2007. 26, 155. DOI: 10.1002/prs.10194
- (a) Speier, J. L.; Webster, J. A.; Bernes, G. H. J. Am. Chem. Soc. 1957, 79, 974. DOI:10.1021/ja01561a054 (b) Lewis, L. N.;Sy, K. G.; Bryant, G. L.; Donahue, P. E. Organometallics 1991,10, 3750. DOI: 10.1021/om00056a055
- Yoshida, J.; Tamao, K.; Takahashi, M.; Kumada, M. Tetrahedron Lett. 1978, 19, 2161. DOI: 10.1016/S0040-4039(01)86834-9
- (a) Ojima, I.; Kumagai, M. J. Organomet. Chem. 1974, 66, C14. DOI: 10.1016/S0022-328X(00)93873-7 (b) Dickers, H. M.; Haszeldine, R. N.; Mather, A. P.; Parish, R. V. J. Organomet. Chem. 1978, 161, 91. DOI: 10.1016/S0022-328X(00)80914-6
- Esteruelas, M. A.; Herrero, J.; Oro, L. A. Organometallics 1993, 12, 2377. DOI: 10.1021/om00030a057
- Oertle, K.; Wetter, H. Tetrahedron Lett. 1985, 26, 5511. DOI: 10.1016/S0040-4039(01)80873-X
- Simonneau, A.; Oestreich, M. Angew. Chem., Int. Ed. 2013, 52, 11905. DOI: 10.1002/anie.201305584
- (a) Rubin, M.; Schwier, T.; Gevorgyan, V. J. Org. Chem. 2002, 67, 1936. DOI: 10.1021/jo016279z (b) Rendler, S.; Oestreuch, M. Angew. Chem., Int. Ed. 2008, 47, 5997. DOI: 10.1002/anie.200801675 (c) Houghton, A. Y.; Hurmalainen, J.; Mansikkamäki, A.; Piers, W. E.; Tuononen, H. M. Nature Chem.2014, 6, 983. DOI: 10.1038/nchem.2063
- Keess, S.; Simonneau, A.; Oestreich, M. Organometallics 2015, 34, 790. DOI: 10.1021/om501284a
- Simonneau, A.; Oestreich, M.;Nature Chem. 2015, ASAP. DOI: 10.1038/nchem.2329
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载