
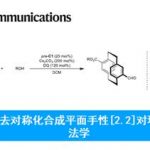
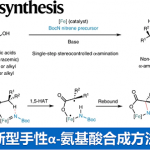
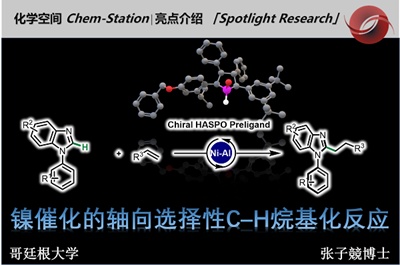
β-手性芳烃化合物广泛存在于具有生物活性的药物和天然产物中(Figure 1A)。传统的合成该类化合物的方法包括两个具有挑战性的步骤:通过不对称合成方法得到的β-手性有机金属试剂再与芳基亲电试剂发生亲电交叉偶联反应。而β-手性有机金属试剂的合成方法包括:(1)卤代烷与强还原性金属或卤化锂发生转金属化反应,然后在Sn、B或Zn等金属作用下再发生转金属化反应[1](Figure 1B),但卤代烷较难获得;(2)利用易得的1,1-二取代烯烃的不对称加氢金属化反应[2],但1,1-二取代烯烃的立体选择性较难控制,而关于1,1-二取代烯烃的硼氢化反应的报道只有三例(Figure 1B)。在上述背景研究的基础上,美国马萨诸塞州麻省理工学院Buchwald课题组 (化学空间: Buchwald教授介绍)报道了钯催化剂和氢化铜催化剂协同催化1,1-二取代烯烃的不对称反马氏规则氢芳基化反应,能以良好的收率和优秀的对映选择性得到一系列β-手性芳烃化合物(Figure 1C)。该反应是有效的一步反应过程,成功解决了多步反应中存在的问题。相关研究成果发表于
“The Enantioselective Preparation of Arenes with β-Stereogenic Centers: Confronting the 1,1-Disubstituted Olefin Problem Using CuH/Pd Cooperative Catalysis”
Lu, Z.; Buchwald, S. L.* Angew. Chem. Int. Ed. 2020, Early View. DOI: 10.1002/anie.202004414
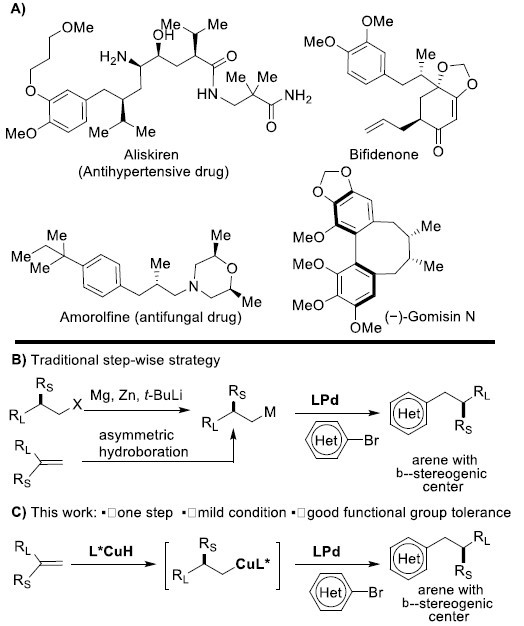
Figure 1 背景研究
论文概要
作者推测可能的反应机理(Figure 2):在Cu催化循环中,铜盐、手性配体和硅烷原位生成CuH催化剂I。然后,1,1-二取代烯烃II发生不对称氢化反应、经历反-马氏规则区域选择性的过渡态III,得到含β手性中心的Cu(I)烷基中间体IV。在Pd催化循环中,Pd(0)络合物V与芳基溴化物发生氧化加成反应生成芳基钯中间体VII。Cu(I)烷基中间体IV与钯中间体VII发生转金属化反应生成β-手性钯烷基络合物VIII和L*CuBr(X)。VIII发生还原消除反应生成不对称β-手性芳烃IX,而Pd(0)中间体V再生进入下一次循环。同时,在碱和硅烷存在下,中间体X可转化为CuH催化剂I。作者必须严格控制这两种催化循环的速率才能得到交叉偶联反应产物。
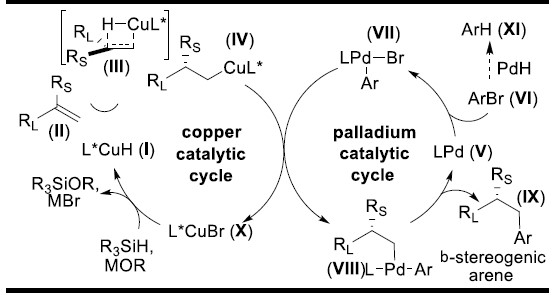
Figure 2 反应机理
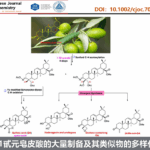
以叔丁基二甲基(3-甲基-2-亚甲基丁氧基)硅烷2a和对甲氧基芳基溴化物为模板底物,作者对催化剂、碱、添加物等反应条件反复筛选,确定最优反应条件:8 mol% Cu(OAc)2和8.8 mol% (R)-L1为Cu催化剂,2 mol%μ-dimer和4 mol% (R)-L1为Pd催化剂,4 equiv CsOBz为碱,2 equiv DMMS为添加物,THF为溶剂,在45℃条件下反应16小时后,加入1.5 equiv TBAF,再在在45℃条件下反应10小时,能以61%的收率和90%的对映选择性得到产物3a。
在最优反应条件下,作者考察了芳基溴化物的底物范围。各种吸电子取代、酯基取代、吡啶、喹啉、噻吩、咔唑、嘧啶等杂环取代的苯基溴化物能较好地适合反应条件,能以良好的收率和优秀的对映选择性得到相应产物。但3-溴喹啉只能以较低的收率和优秀的对映选择性得到相应产物。
紧接着,作者考察了烯烃的底物范围(Table 1)。无论是小位阻取代的烯烃还是大位阻取代的烯烃均能较好的适应反应条件,能以良好的收率和优秀的对映选择性得到相应产物。但无TBS保护的烯烃只能以较低的收率和优秀的对映选择性得到相应产物。
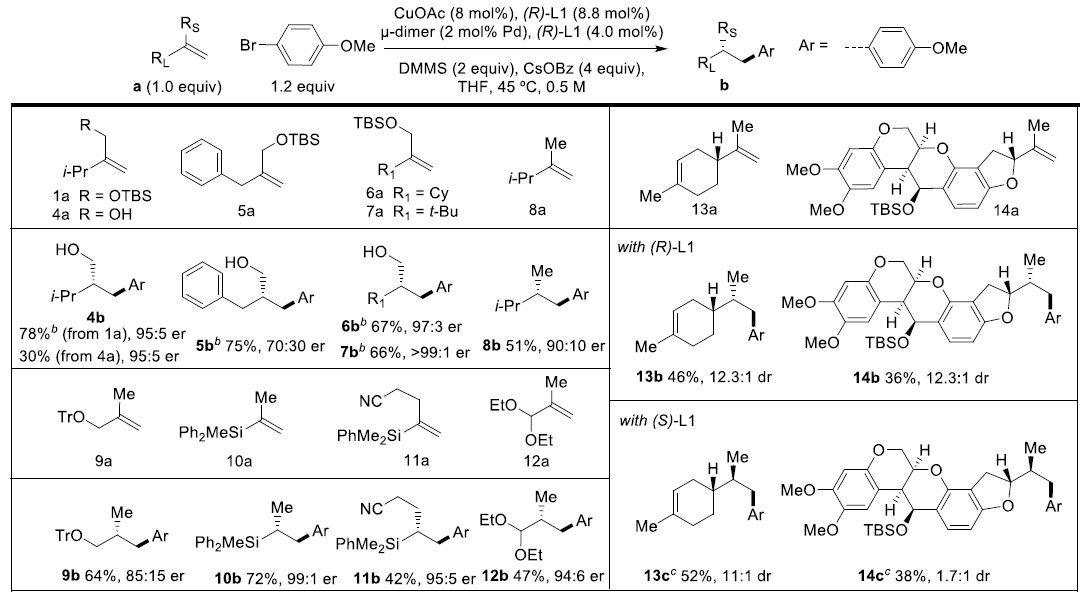
Table 1 底物扩展
论文总结评价
美国马萨诸塞州麻省理工学院Buchwald课题组报道了钯催化剂和氢化铜催化剂协同催化1,1-二取代烯烃的不对称反马氏规则氢芳基化反应,能以良好的收率和优秀的对映选择性得到一系列β-手性芳烃化合物。该反应是有效的一步反应过程,成功解决了多步反应中存在的问题。
参考文献
- [1] Chemler, S. R.; Trauner, D.; Danishefsky, S. J. Angew. Chem. Int. Ed. 2001, 40, 4544-4568. DOI: 10.1002/1521-3773
- [2] Jang, W. J.; Song, S. M.; Moon, J. H.; Lee, J. Y.; Yun, J. J. Am. Chem. Soc. 2017, 139, 13660-13663. DOI: 10.1021/jacs.7b08379
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!














No comments yet.