

本文作者:芃洋雪
有机电合成
相比于化学试剂,电能可以直接转化为化学电位。非均相的电子可以在固相的电极和溶液的界面上传递。此时,溶液分子本身或者电解质与溶液中的反应底物一起参与到电子转移过程。
电化学合成其实历史悠久,早在19世纪就用电解制备大宗化学品,但相比之下,实验室电化学的发展要远远落下了,直至最近有机电合成才逐渐兴起,其复兴归功于在制备复杂产物时的高效率、高选择性,以及高原子经济性、安全性。并且随着技术和设备的发展,电化学方法更加方便。下面介绍几个电化学合成的实验案例,体会其独特的魅力。
金属催化电合成邻二胺
相比与烯烃的二醇化反应,烯烃的二胺化反应还需完善。电化学方法可以将烯烃22转化为1,2-二叠氮物23,再还原为1,2-二胺24。将过渡态金属催化和电合成组合,反应条件温和,应用范围广,底物包括大位阻和多取代的烯烃,如1,1-二取代,1,2-二取代,三取代和四取代的底物都可以用于这个反应方法学,具有优秀的化学选择性和官能团兼容性,如图a所示。其反应机理可能为阳极氧化叠氮为自由基,然后加成到双键上,氧化还原活性的Mn催化剂介导了反应保证自由基25生成邻二叠氮化物23。通过精准控制电压,阳极电位正好满足氧化反应的最低电位,因此反应具有高度的化学选择性,没有引起其他副反应[1,2]。
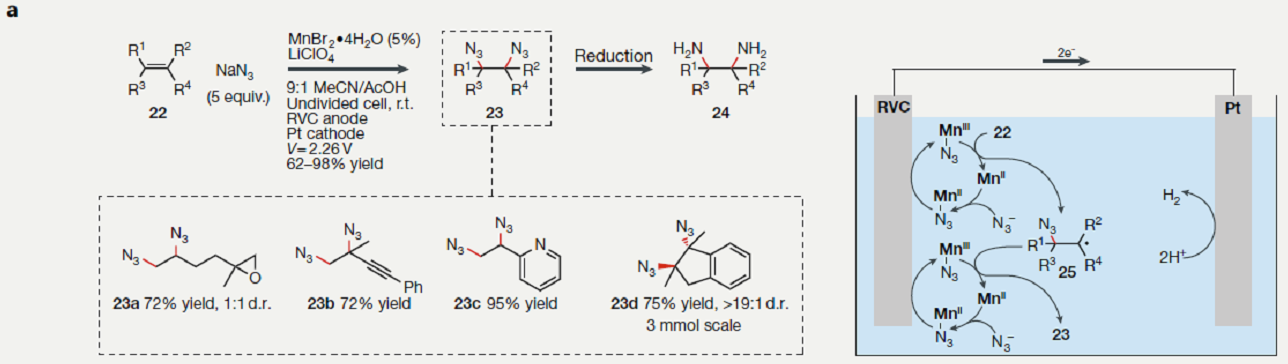
图a. Mn电极催化的烯烃二叠氮化反应,具有优秀的化学选择性和广泛的应用范围.
图片来源:Nature Reviews Chemistry
阳极不对称芳基偶联反应
C-H键的直接官能团化反应是目前有机化学的活跃领域之一,吸引了众多形式的合成方法,电化学合成也是其中之一。电化学条件下苯酚26和硅保护的苯酚衍生物27可以一步反应完成对称的C-C偶联,图b所示[3]。硼掺杂金刚石boron-doped diamond(BDD)阳极和1,1,1,3,3,3-六氟丙醇(HFIP)作为H-键受体溶剂是合成邻位偶联产物的关键。硅SiiPr3保护基团对27的电子效应有微弱影响,同时防止产物28继续氧化。反应条件优化后,最佳反应为50 oC,之前虽然也有电化学反应的芳基偶联反应报道,但选择性很差,常生成未保护苯酚低聚反应。与依靠化学试剂,如化学计量的氧化剂等进行的芳基偶联方法相比,这个电合成方法更胜一筹。

图b. 无金属和配体试剂的C-C偶联反应,选择性制备保护的不对称2,2′-联苯二酚和2,2′-联苯二胺,HFIP是强H-键受体,起到稳定自由基中间体的重要作用.
图片来源:Nature Reviews Chemistry
与H2O或甲醇相比,溶剂HFIP更倾向于将亲核性和氧化还原电位区分开来,虽然它的具体作用还未知,但和它具有高度的氧化还原稳定性、H-键作用、增强酸性、减少亲核性等性质相关,从而稳定了电化学合成中的自由基中间体。它还应用在酰胺或氨基甲酸酯保护的胺29和30反应生成2,2-二氨基联苯产物31,收率为中等至优秀[4]。这个方法具有普适性,苯酚可以和其他芳香杂环,如噻吩、呋喃和苯并呋喃偶联[5]。
有机电合成中稳定“阳离子池”
电化学氧化常常产生碳正离子,它容易被亲核试剂捕获。然而,在未分离的电化学反应池中,碳正离子会被阴极还原,而亲核试剂在阳极氧化,使反应更复杂,从而阻碍了其发展。为解决这个问题,阳离子池策略随之诞生,用分离的反应池从物理空间上分开阳离子产生和亲核试剂捕获,反应通过两步反应进行。典型情况下,阳极电解质中发生碳正离子产生和富集,然后向这个混合物物中加入亲核试剂,在非氧化条件下捕获活泼中间体。例如阳离子捕获剂硫亚胺33和阳极产生的碳正离子40反应,生成亚稳的苄基氨基锍盐衍生物34[6]。这种策略,可以有效生成阳离子,稳定了活泼的中间体,34可以和碳亲核试剂如苯衍生物、芳香杂环、烯醇醚类、烷基三甲基硅烷、NBu4I等反应,得到目标产物35-39[7]。
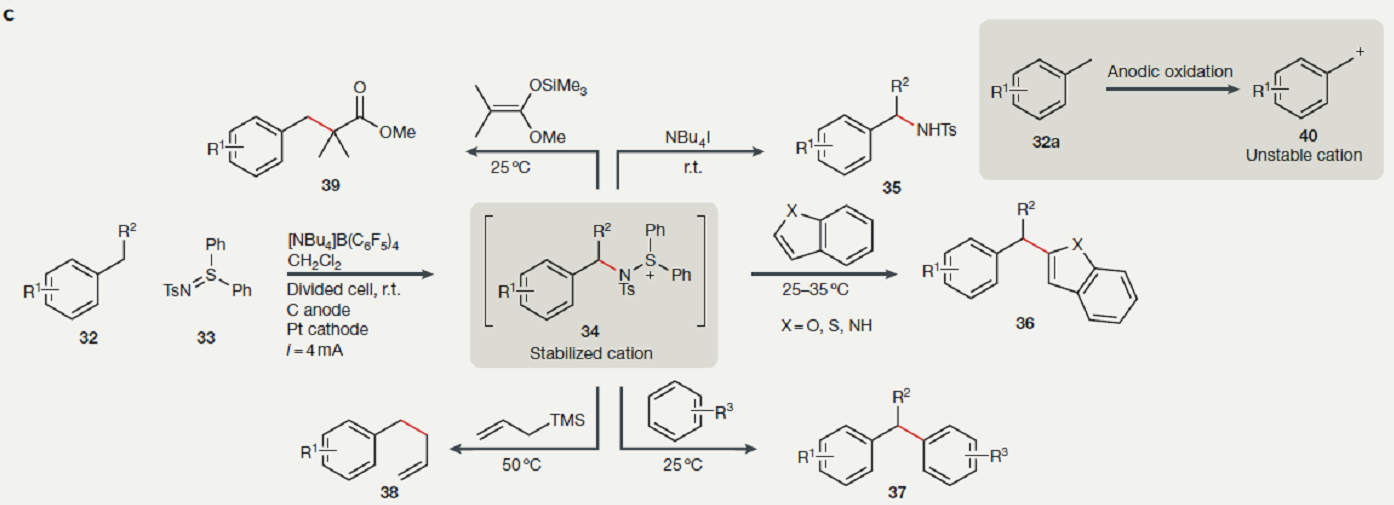
图c. 电产生的稳定的“阳离子池”,阳离子寿命够长,足以在下阶段和亲核试剂反应.
图片来源:Nature Reviews Chemistry
电化学反应器的发展
过去电化学合成属于小众研究,因此于缺乏标准设备和材料,以及简便的操作,这阻碍了有机电合成发展,一些化学家不得不自制装备进行研究。现在简单或特定制作的设备打破了这些障碍,学术界和制造商合作,有可能推出适合实验室规模的设备,并且配套的电极,反应池及其他配件有助于发展。
静电场化学
静电场化学容易和电合成混淆,静电其实也可以作为催化剂。众所周知,大部分分子以及反应过渡态都有极性,理论上通过加入合适的电场,可以稳定在某种状态,而近年来的几个实验也证实了这种设想。
单电子实验
第一个外加电场催化的反应是本综述的作者开发的Diles-Alder反应,实验使用了扫描隧道显微镜STM。通过巯基将降冰片烯衍生物固定在STM的金片表面上,呋喃衍生物固定在探针上这样,这些分子的朝向已知并且被控,并且距离固定,如电压恒定则试剂所在的电场强度固定。显微镜开启在blinking模式,以恒定的电压变化速率扫描。二烯和亲二烯体反应时,新生的共价键形成部分在探针和金片之间,并形成一次电流。通过计数每单元时间的电流”blinks”,可以测量反应速率。当是电场是负时,D-A反应的速率随电场强度的增加而提高,相对于相同条件下电场为0时速率提高了5倍。当电场为负时,反应速率保持在最低水平。即只有当电场的极性促进电子从亲二烯体分子流向二烯分子时,反应速率才能得到提高, 反应方法如图d所示[8]。
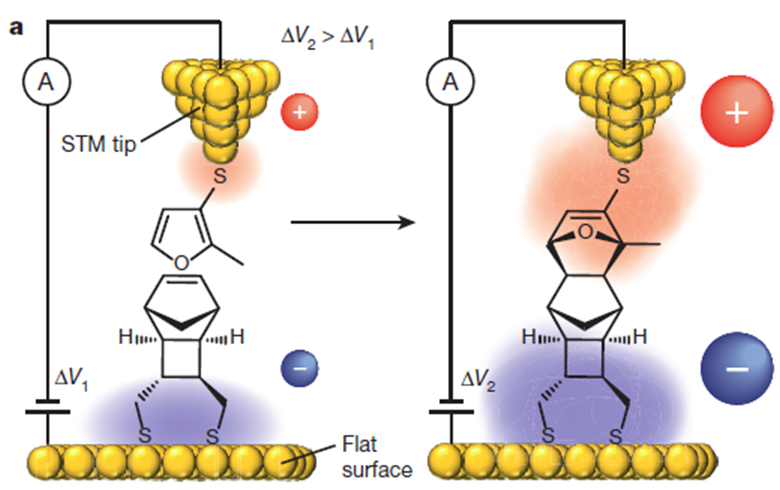
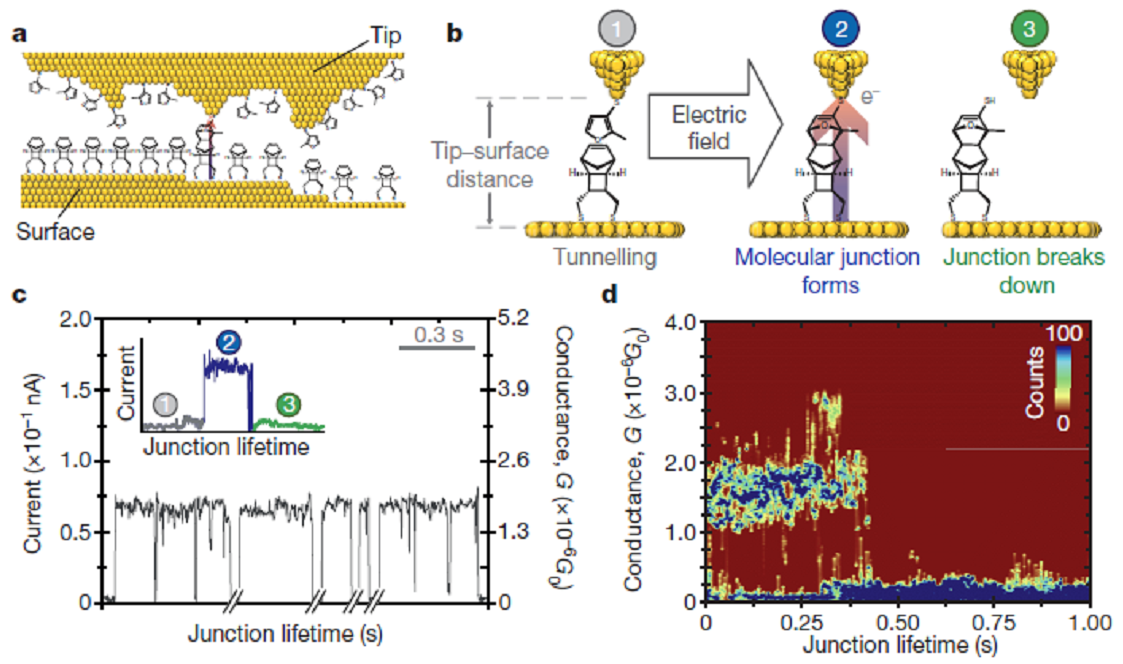
图d. 图片来源:Nature 2016, 531, 88-91.
静电场化学还可以扳动化学键的断裂,如烷氧基胺分解为氧化氮和碳自由基[9]。一般而言,单分子反应为研究静电场对化学反应的影响提供了良好的平台,也扩展了纳米技术应用如显微成图。但这个方法不适用于大规模合成。
界面实验
在电化学反应池中,因形成电子双层结构,电极表面的电场为(~0.5Vnm-1),这个场强和STM实验类似,有可能以较大规模催化化学反应。作为一种概念验证,电化学氧化烷氧基胺,快速可逆断裂为氧化氮自由基和碳正离子,如下图e所示,虽然这看上去很像电化学合成,实际反应只有电子双层可以达到[10]。
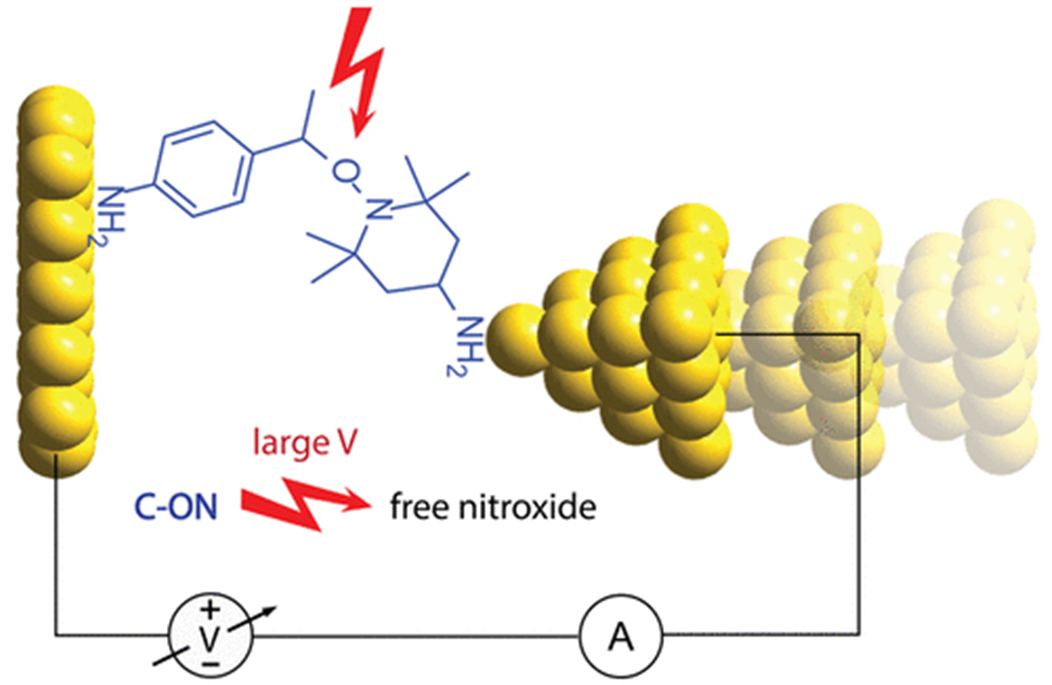
图e. 图片来源 J. Am. Chem. Soc., 2018, 140, 2, 766-774.
另外一个案例是阴离子-催化剂,在氧化铟锡电极的电场作用下,烯醇加成到丙二酸半硫酯上,活性提高了100多倍,如图f所示[11]。
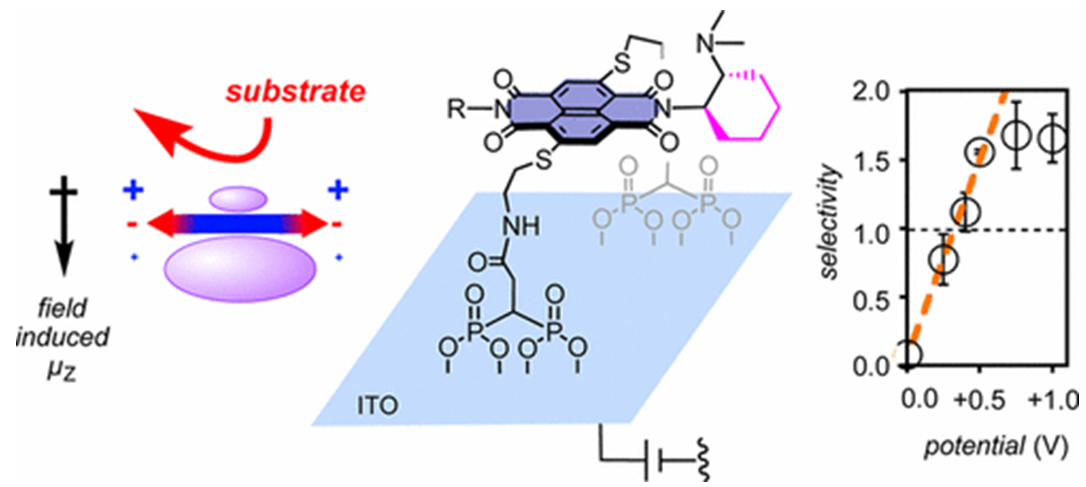
图f. 图片来源 J. Am. Chem. Soc., 2017, 139, 6558-6561.
另外一种方式是使用平行板电池,在标准电极上面隔离出一层膜。施加电压后,表面带有电荷,从而可以催化或提高化学反应的选择性。如顺式二苯乙烯氧化物的重排反应,在电场作用下,醛:酮产物的比例提高至63,如图g所示[12]。
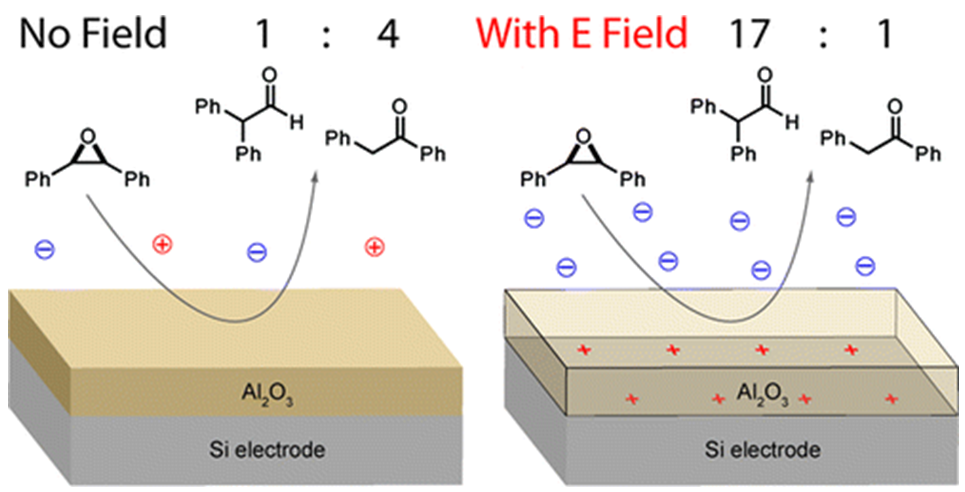
图g. 图片来源:J. Am. Chem. Soc., 2012, 134, 186-189.
Rh催化剂催化1-diazo-3,3-dimethyl-5-phenylhex-5-en-2one 1-Diazo-3,3-dimethyl-5-phenylhex-5-en-2-one的分子内重排反应,无电场下,环丙烷产物和环己酮产物的比例约为10:1,有电场条件下,不同条件下、产物比例可达到为100:1和1:2,如图h所示[13]。
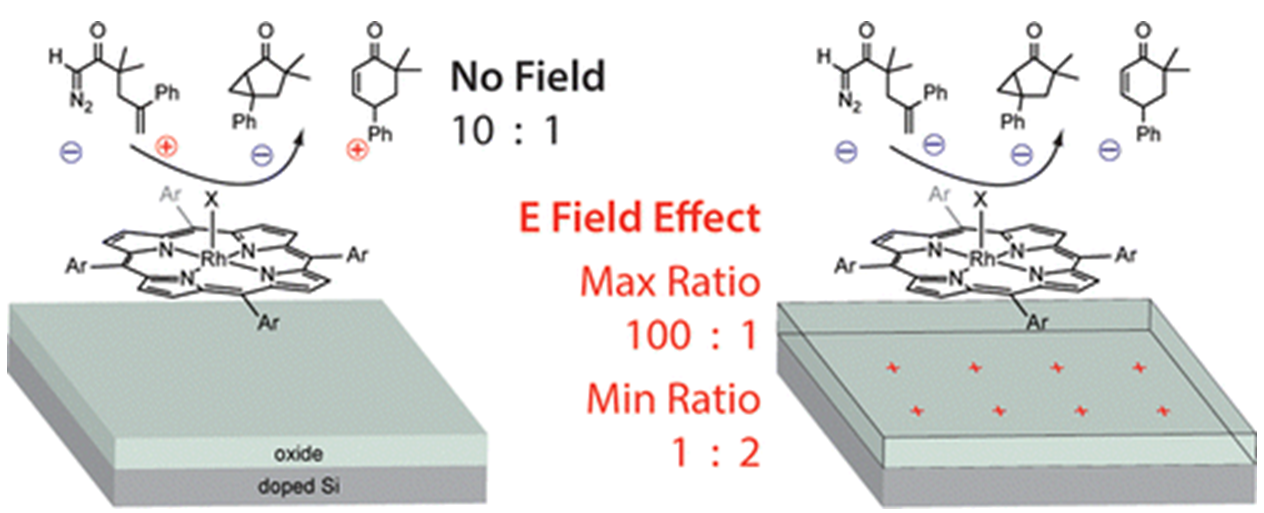
图h. 图片来源:J. Am. Chem. Soc.2013, 135, 11257-11265.
官能团的电荷化
另外一种真正规模化化学合成的方式是将官能团电荷化后的电场如Brønsted酸或碱,金属离子(簇)。当使用Bronsted酸或碱时,静电效应被简单替换为改变pH。电荷基团的静电场作用距离很短(∝1/r2,电荷-偶极作用反比于距离的平方),依靠电荷基团仅能引起分子或超分子的自组装。但将电荷基团精准摆放,使其朝向一致,就不需要表面化学技术。在强电介质中,静电场效应被削弱,因此反应条件一般在气相或非极性溶剂中,挥发性和溶解性是重要的参数。尽管如此,也可以在溶液中观察到了强大的静电效应。例如底物中的极性电荷基团,加速了它的D-A反应,改变了反应的立体选择性和非对映选择性,反应案例图i。2-吡喃酮2-pyrone和环戊烯cyclopentene衍生物的D-A反应,取代基有胺或羧基. 如果环戊烯为中性,反应经过没有位阻的exo过渡态(C),如为胺基被质子化,反应经过有位阻的exo过渡态(D).当羧酸去质子后,反应经过有位阻的endo过渡态(B)。但不论哪种取代基,反应能障都低于中性形式。在电子云密度图上可以看出,变化原因为电荷基团稳定了反应的偶极过渡态[14]。


图i. 静电作用应用于有机合成. 图片来源:Nature Reviews Chemistry
极性和溶解性之间的矛盾,可以通过非均相催化剂调和。下面是一例早期的反应,静电场起到了重要作用。早在1970年,就有LiClO4催化催化tBuCl的HCl消除反应和1-苯基烯丙基氯的重排反应,直至最近这种提高反应速率或改变反应选择性的电场效应才被理解。最近,值得关注的研究领域是气相中CH4和H2的活化,[TaO2]+电荷复合物起到了电催化的作用,图j所示[15]。
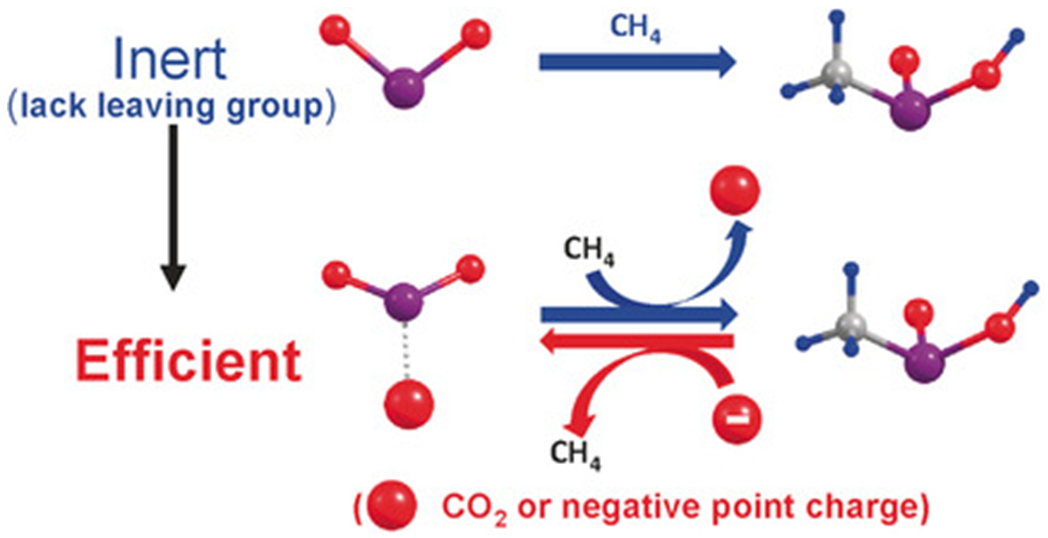
图j. 图片来源: Angew. Chem. Int. Ed., 57, 14635
还有自由基化学,负电荷相比中性物质可以更稳定双自由基,可以用来开发pH控制的自由基聚合反应和自由基生物化学,电场作用可以加强路易斯酸催化自由基加成反应,或者可以帮助解释离子液体的在化学反应中的作用[16-21]。
参考文献
- Schäfer,, H. Oxidative addition of the azide ion to olefins. A simple route to diamines. Angew. Chem. Int. Ed. 9, 158–159 (1970).
- Fu, N., Sauer, G., Saha, A., Loo, A. & Lin, S. Metal-catalyzed electrochemical diazidation of alkenes. Science 357, 575–579 (2017).
- Wiebe, A., Schollmeyer, D., Dyballa, K. M., Franke, R. & Waldvogel, S. R. Selective synthesis of partially protected nonsymmetric biphenols by reagent- and metal-free anodic cross-coupling reaction. Angew. Chem. Int. Ed. 55, 11801–11805 (2016).
- Schulz, L. et al. Reagent- and metal-free anodic C–C cross-coupling of aniline derivatives. Angew. Chem. Int. Ed. 56, 4877–4881 (2017).
- Kärkäs, M. D. Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 47, 5786–5865 (2018).
- Hayashi, R., Shimizu, A. Yoshida, J.-i. The Stabilized Cation Pool Method: Metal- and Oxidant-Free Benzylic C–H/Aromatic C–H Cross-Coupling. J. Am. Chem. Soc. 138, 8400–8403 (2016).
- Hayashi, R. et al. Metal-free benzylic C-H amination via electrochemically generated benzylaminosulfonium ions. Chem. Eur. J. 23, 61–64 (2017).
- Aragonès, A. C. et al. Electrostatic catalysis of a Diels–Alder reaction. Nature 531, 88–91 (2016).
- Zhang, L. et al. Electrochemical and electrostatic cleavage of alkoxyamines. J. Am. Chem. Soc. 140, 766–774 (2018).
- Zhang, L. et al. Electrochemical and electrostatic cleavage of alkoxyamines. J. Am. Chem. Soc. 140, 766–774 (2018).
- Akamatsu, M., Sakai, N. & Matile, S. Electric-field-assisted anion-π catalysis. J. Am. Chem. Soc. 139, 6558–6561 (2017).
- Gorin, C. F., Beh, E. S. & Kanan, M. W. An electric field-induced change in the selectivity of a metal oxide?catalyzed epoxide rearrangement. J. Am. Chem. Soc. 134, 186–189 (2012).
- Gorin, C. F., Beh, E. S., Bui, Q. M., Dick, G. R., Kanan, M. W. Interfacial electric field effects on a carbene reaction catalyzed by Rh porphyrins. J. Am. Chem. Soc. 135, 11257–11265 (2013).
- Aitken, H. M., Coote, M. L. Can electrostatic catalysis of Diels–Alder reactions be harnessed with pH-switchable charged functional groups?Phys. Chem. Chem. Phys. 20, 10671–10676 (2018).
- Yue, L. et al. The electric field as a “smart” ligand in controlling the thermal activation of methane and molecular hydrogen. Angew.Chem. Int. Ed. 57, 14635–14639 (2018).
- Gryn’ova, G., Coote, M. L. Origin and scope of long-range stabilizing interactions and associated SOMO–HOMO conversion in distonic radical anions. J. Am. Chem. Soc. 135, 15392–15403 (2013).
- Gryn’ova, G., Marshall, D. L., Blanksby, S. J. & Coote, M. L. Switching radical stability by pH-induced orbital conversion. Nat. Chem. 5, 474–481 (2013).
- Gryn’ova, G., Smith, L. M., Coote, M. L. Computational design of pH-switchable control agents for nitroxide mediated polymerization. Phys. Chem. Chem. Phys. 19, 22678–22683 (2017).
- Jiang, J. Y., Smith, L. M., Tyrell, J. H., Coote, M. L. Pulsed laser polymerisation studies of methyl methacrylate in the presence of AlCl3 and ZnCl2 — evidence of propagation catalysis. Polym. Chem. 8, 5948–5953 (2017).
- Clark, T. Lithium cation as radical-polymerization catalyst. J. Am. Chem. Soc. 128, 11278–11285 (2006).
- Vyakaranam, K., Barbour, J. B. & Michl, J. Li+-catalyzed radical polymerization of simple terminal alkenes. J. Am. Chem. Soc. 128, 5610–5611 (2006).
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!




















No comments yet.