
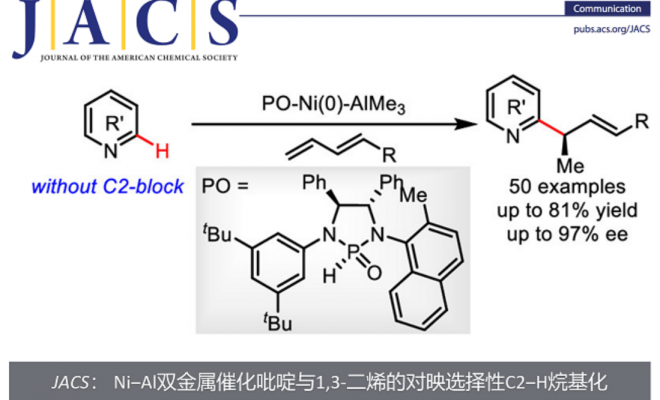
作者:石油醚
导 读
近日,南开大学叶萌春课题组使用二胺衍生的膦手性膦氧配体桥联镍-铝双金属催化剂实现无C2-取代基位阻屏蔽的吡啶C2‒H与二烯分子间的不对称烷基化反应,相关成果发表在J. Am. Chem. Soc.上 (https://doi.org/10.1021/jacs.2c09306)。
“Enantioselective C2–H Alkylation of Pyridines with 1,3-Dienes via Ni–Al Bimetallic Catalysis.
Jiang-Fei Li, Deng Pan, Hao-Rui Wang, Tao Zhang, Yi Li, Genping Huang*, and Mengchun Ye*
J. Am. Chem. Soc.2022, ASAP. doi: 10.1021/jacs.2c09306”
正 文
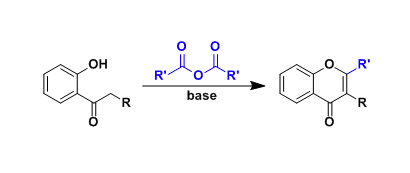
C-2手性烷基取代的吡啶广泛存在于医药,农药和生物活性天然产物中,同时,又可以作为手性催化剂催化有机反应。因此,它们的合成具有重要的研究意义和应用价值(图 1a)。
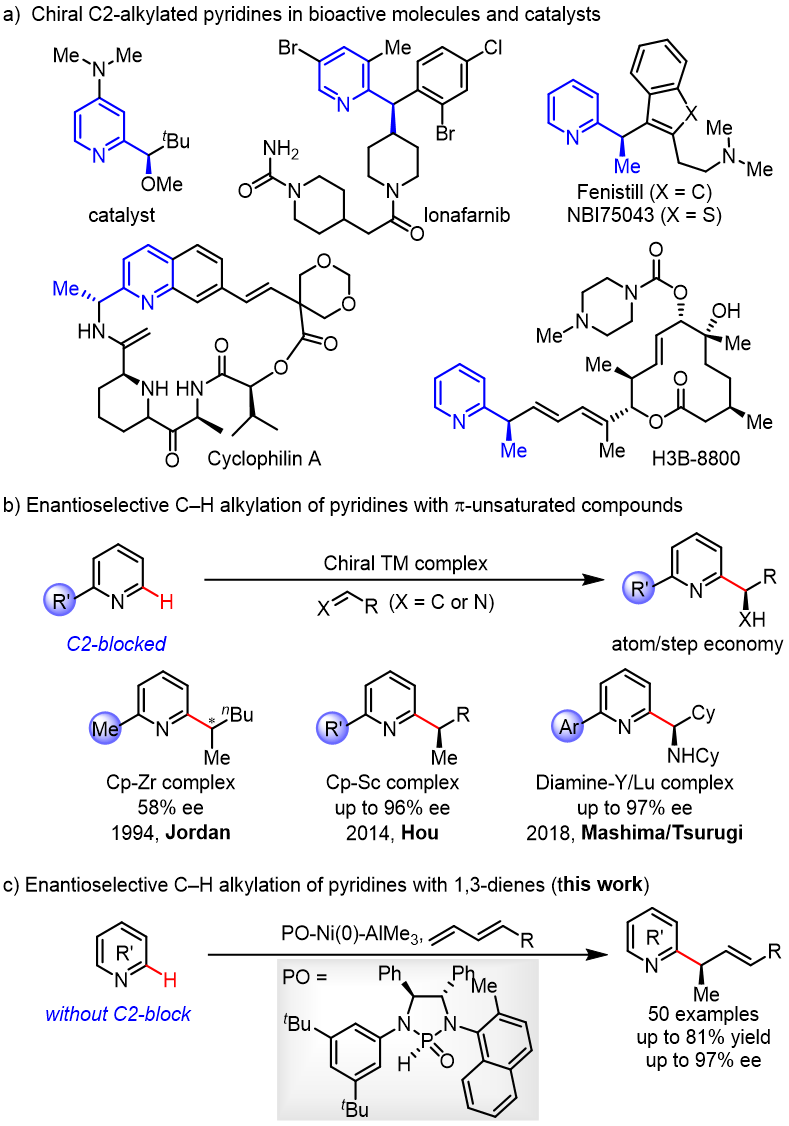
图1. Ni‒Al双金属催化吡啶与1,3-二烯的对映选择性C2‒H烷基化。
目前,合成C-2手性烷基化吡啶的方法主要有两类,一类是使用预官能化的吡啶和当量的手性试剂;另一类是吡啶的Minsci反应,但常常需要牺牲当量的分子结构碎片,降低了反应的原子经济性。通过过渡金属催化的吡啶C2‒H键与π-不饱和化合物加成反应是构建此类化合物最为直接和经济的方法,不仅不需要预官能化的吡啶和特殊的烷基化试剂,而且反应具有完全的原子经济性。然而,由于吡啶对金属的强配位能力,抑制了手性配体对金属的配位,这类反应的立体选择性控制一直是一个较大的挑战,成功的例子较为罕见。1994年,Jordan课题组使用手性Cp‒Zr复合物首次实现了2-己烯与C2-取代吡啶的C2‒H烷基化反应,获得58%ee(图 1b)。 2014年,Hou课题组使用阳离子Sc复合物实现了C2-取代吡啶的C6‒H烷基化反应,获得高达96%的ee。 2018年,Mashima和Tsurugi课题组使用手性二胺的Y或Lu复合物催化了C2-芳基吡啶的C-H氨基烷基化反应,获得高达97%的ee(图 1b)。尽管已经取得了较大进展,但所有这些方法都需要使用C2取代的吡啶,通过C2位取代基的位阻效应来抑制吡啶与金属的配位。然而这一需求导致底物和产物结构的严重受限,致使广泛存在的非C2取代吡啶分子无法兼容这些方法。为了解决这一挑战,南开大学叶萌春课题组利用PO/Ni/Al双金属协同催化体系,实现了无C2取代基位阻屏蔽的吡啶与烯烃的不对称烷基化反应,合成了一系列手性吡啶类衍生物,获得45–82%的收率和高达97% ee(图 1c)。
作者选择简单吡啶1a和苯基1,3-二烯2a作为模板底物,对手性磷氧配体进行了考察(图2)。当膦手性膦氧配体L15作为反应配体,AlMe3作为路易斯酸,在135 ℃条件下,可得到82%的收率和94%的ee值。通过单晶X射线衍射分析,确定产物主要对映体的构型为(R)构型。

图2. 反应条件优化
随后作者对吡啶的底物范围进行考察(图3)。结果显示,各种C2,C3-或C4-取代基都能得到较高的收率和ee值,其中C2位位阻较大时会影响Al与吡啶的配位,从而影响反应的发生。此外。喹啉(3w)和喹啉(3x)也能兼容该反应。
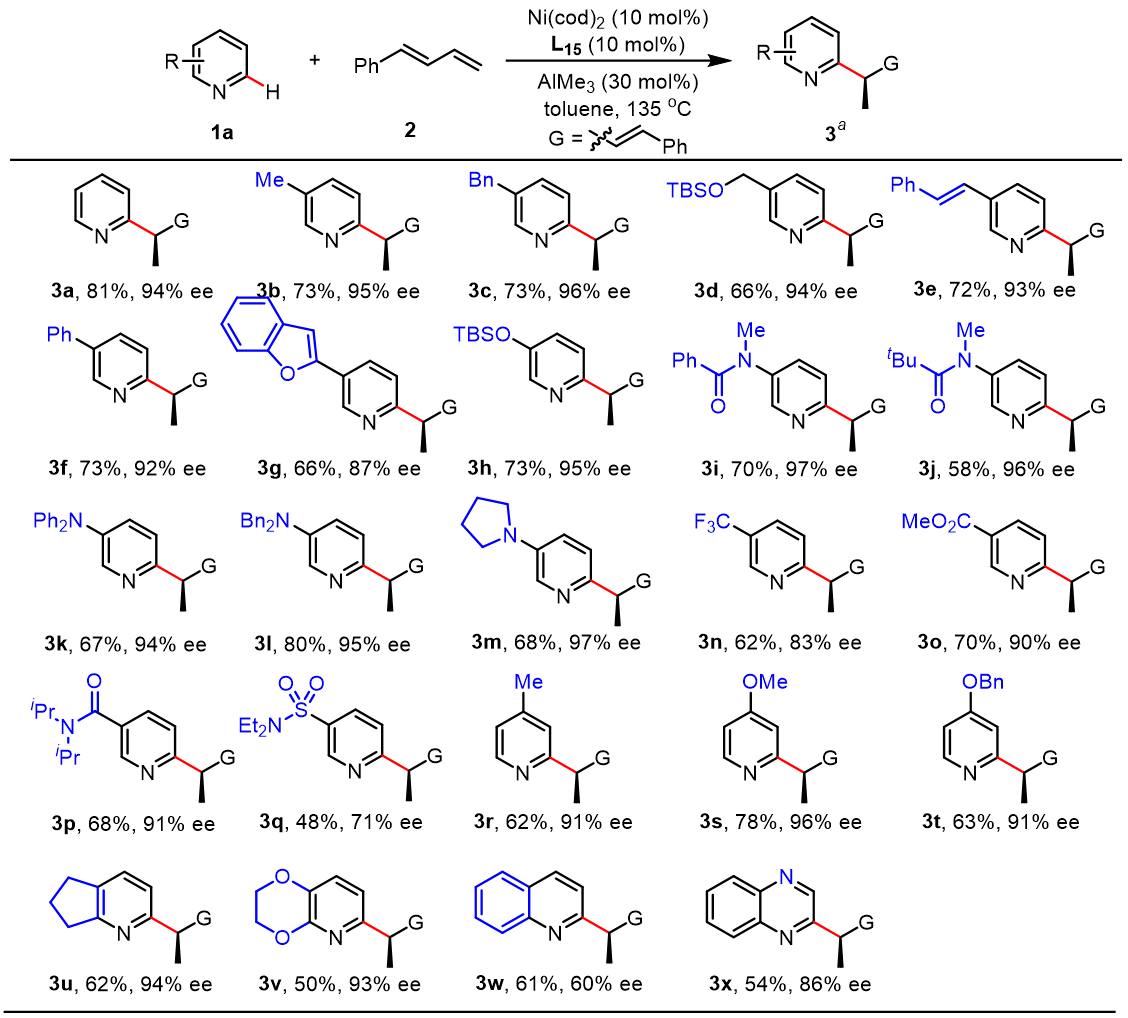
图3. 底物拓展-吡啶
接下来作者考察了1,3-二烯的范围(图4)。对于芳基二烯,吸电子和电子取代基都具有较好的兼容性,除了单取代1,3-二烯外,二取代1,3-二烯也能顺利参与反应,得到相应的产物。
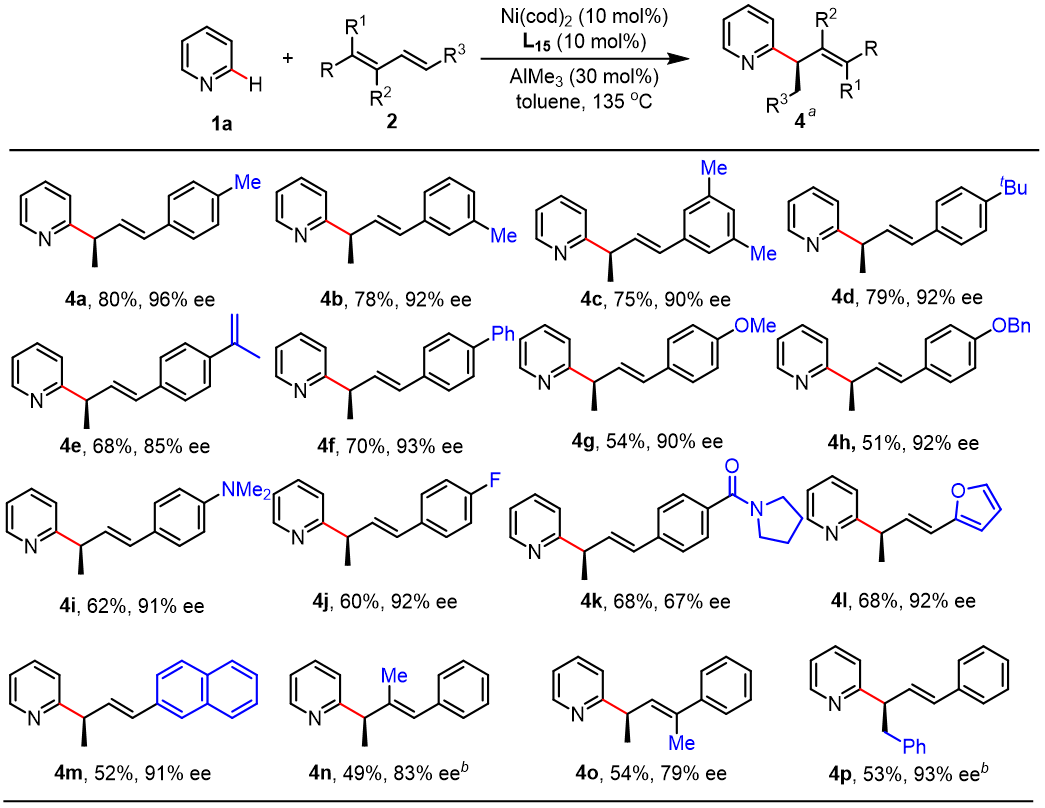
图4. 底物拓展-1,3-二烯
同时作者将这种方法应用于含吡啶的生物活性复合物分子和有机催化剂的后期修饰 (图5)。4-(二甲氨基)吡啶(DMAP)和4-吡咯酰吡啶(PPY)是亲核反应中经典的有机催化剂,但相应的手性催化剂的合成则比较困难,常常需要冗长的反应步骤。然而,使用目前的方法可以一步法合成它们。同时,该方法可以应用于一些生物活性含吡啶的复杂分子的直接修饰。

图5 底物拓展-含吡啶有机催化剂和生物活性分子
为了考察反应的实用性,作者进行了产物转化:双键的还原可以得到手性吡啶6;3a的氧化,随后还原生成醇7 (图6a)。接着作者对反应机理进行了探索,发现吡啶的C2‒D分布在产物的两个位置,包括甲基(1.21 D)和烯丙基氢(0.27 D),同时,C5‒D被H (0.48 H)部分取代(图6b)。这些结果表明烯丙基镍的形成是一个可逆过程,这将导致C5‒D和甲基H的交换。此外,平行实验中没有显示显著的动力学同位素效应(kH/kD = 1.04) (图6c),表明C2−H的断裂不是反应决速步骤。DFT计算结果表明,(1) C2‒H活化通过可逆的配体-配体氢转移途径((R)-TS1)进行; (2) h1和 h3烯丙基镍络合物之间异构化生成中间产物IM4,然后还原消除生成产物3a; (3)还原消除步的能垒为30.2 kcal/mol,暗示还原消除的决速步; (4)计算得到两个立体异构体的路径之间的能量差为2.7 kcal/mol,这与实验结果94% ee值相吻合; (5)对映体选择性主要是由于S途径的C‒C还原消除比R途径的能量高得多。在(R)-TS3中,计算结果烯丙基的苯基与配体的N -苯基之间存在C‒H—π相互作用,在(S)-TS3中,计算结果显示烯丙基与Ni-Al催化剂之间存在位阻效应。此外,DFT计算还排除了吡啶C3‒或C4‒H活化,以及1,3-二烯的位点选择性。
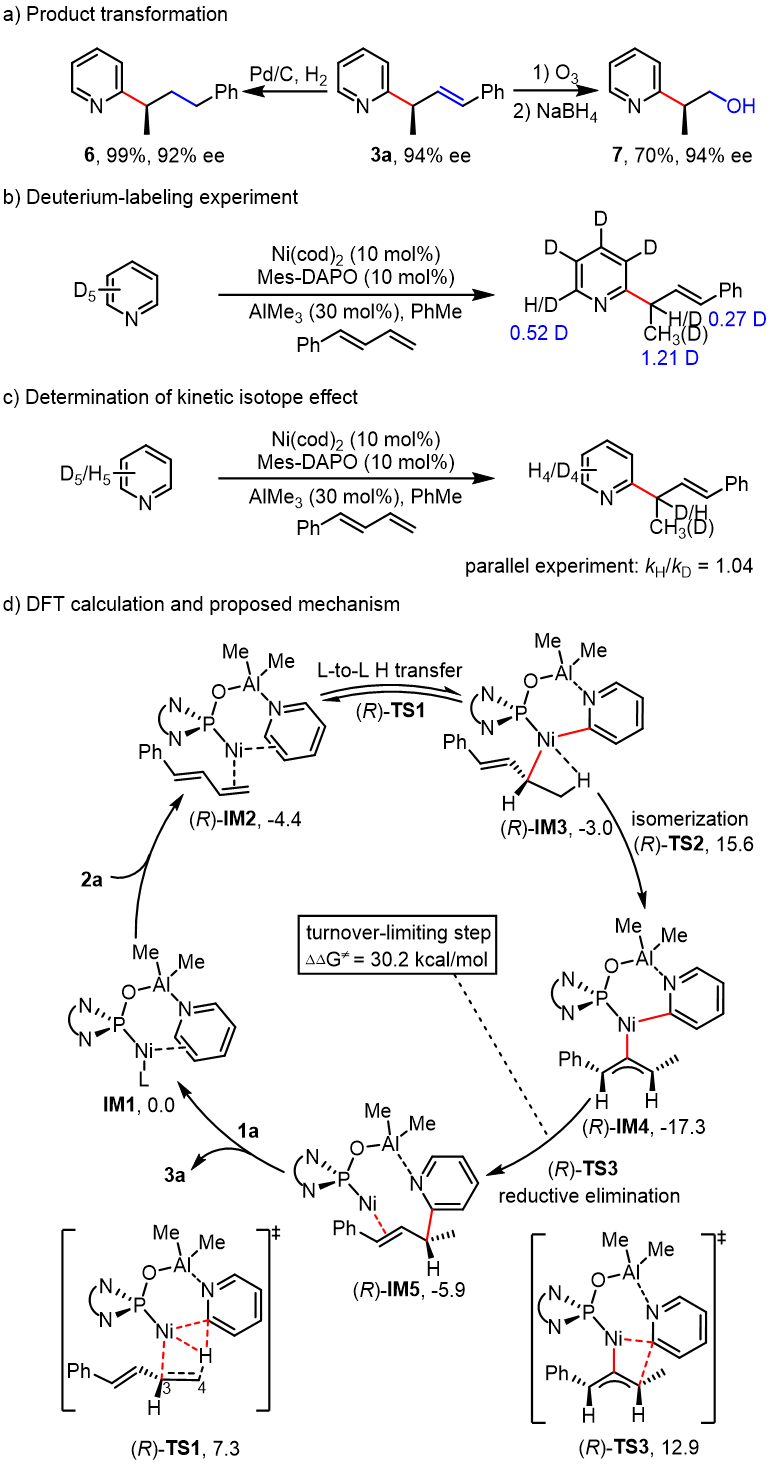
图6 .转化与机理
总 结
南开大学叶萌春课题组使用二胺衍生的膦手性膦氧配体桥联镍-铝双金属催化剂实现了无C2取代基位阻屏蔽的吡啶与烯烃的不对称烷基化反应,合成了一系列手性吡啶类衍生物,获得45–82%的收率和高达97% ee。该反应中,作者发现了新的含膦手性的膦氧配体,获得了较高的立体选择性。该类配体和双金属催化模式有望在更多的反应中得到应用。
(叶萌春教授供稿)
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载

















No comments yet.