
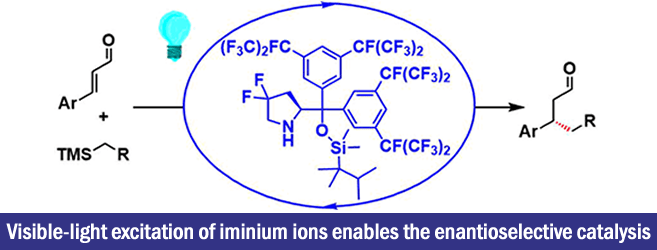
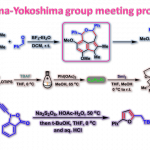
2017年、加泰罗尼亚化学研究所・Paolo Melchiorre课题组、通过用可见光激发亚胺有机催化剂体系发现了该体系具有强氧化能力。利用这一点,实现了一种无法通过热反应进行的不对称催化反应(烷基硅烷氧化生成烷基自由基,然后对烯醇进行不对称1,4-加成)。
“Visible-light excitation of iminium ions enables the enantioselective catalytic β-alkylation of enals”
Silvi, M.; Verrier, C.; Rey, Y. P.; Buzzetti, L.; Melchiorre, P.* Nat. Chem. 2017, 9, 868-873. doi:10.1038/nchem.2748
课题设定
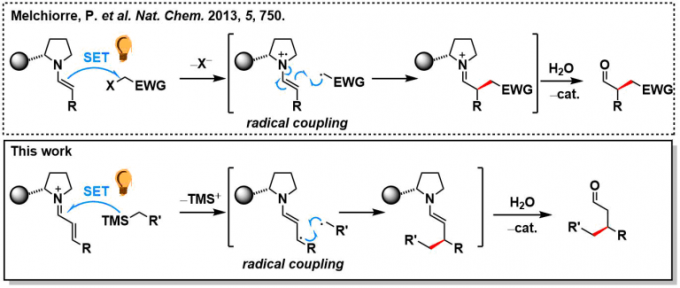
有机催化剂的代表之一亚胺鎓型手性催化剂,在过去几十年来被开发用于各种手性催化反应中。但是,这些反应中,催化剂都是出于基态的状态发挥作用的。通过事先配制的亚胺离子再通过光照等条件变成激发态后用于自由基催化反应的虽然已经有报道[1],但是应用于手性催化领域的目前还很少有实例出现。
解决手法
作者通过使用烯胺催化剂+可见光激发+还原敏感底物的组合、报道了一些醛的可见光下驱动的不对称α―烷基化反应[2]。以此为启发,作者这次产生了尝试利用亚胺鎓催化剂+可见光激发+氧化敏感底物的组合的想法。
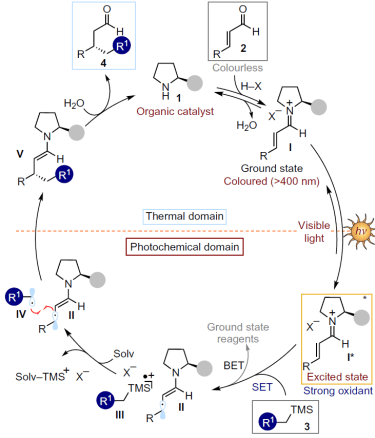
假定的催化剂循环如下所示。通过可见光激发的亚胺激活态I*(π-π*迁移、>400 nm)氧化烷基硅烷3,生成烷基自由基IV、在此之后引起不对称自由基偶联,反应通过烯胺V的水解完成一个循环。
引用自原论文
烷基三甲基硅烷用作了碳自由基的前体,这是因为
- 具有相对低的氧化电位[3]
- 它的自由基阳离子在弱亲核剂(与乙腈相当的)共存在可以快速发生脱硅基化[4],生成烷基自由基,这是不可逆的过程,因此可以回避反向的电子移动。
这两个反应设计上的优点。
主张的有效性验证
① 催化剂结构的筛选
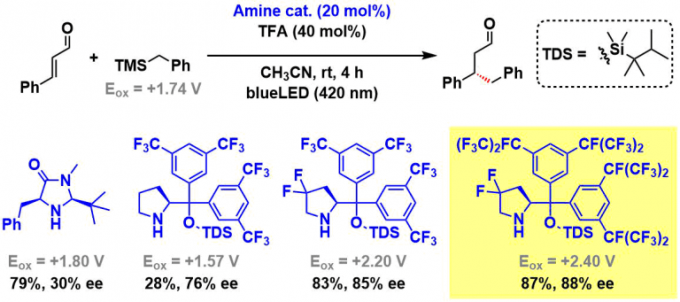
作者先以肉桂醛和苄基TMS为底物进行催化剂筛选。胺催化剂的结构需要激发态亚胺I*具有足够能氧化烷基硅烷(Eox = +1.74V, vs Ag/Ag+)的氧化电位、并且胺基催化剂自身还必须不能还原激发态I*。由于这些必须因素,作者使用了在某种程度上几乎不被氧化的仲胺催化剂。
底物在与MacMillan催化剂(20 mol%)共存下,通过420 nm LED光照射尝试进行反应,发现虽然得到了目标1,4-附加产物(产率79%),但是ee并不理想。另一方面,当基于Hayashi-Jorgensen催化剂骨架进行结构改造时,发现未修饰的吡咯烷催化剂的活性低。这是因为催化剂本身比烷基硅烷更容易被氧化(Eox = +1.57V)、是由于自氧化破坏(NMR实验证实)。最后通过引入氟原子使得催化剂的氧化耐受性提高,同时反映活性跟手性产率也提高。最终以下图黄色标记的催化剂为最优催化剂进行下一步探讨。
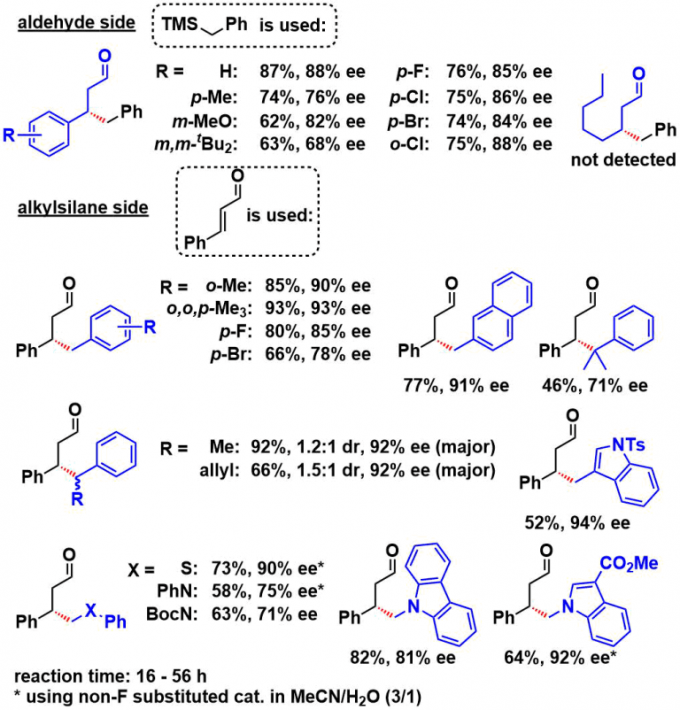
② 底物拓展
首先使用苄基作为亲核剂本身是非常困难的,并且相同形式的对烯酮的不对称1,4-加成的也并没有报道过,这次是首次。
底物的拓展如下所示。
- β-烷基烯酮不适用(极有可能生成的亚胺中间体在没有在可见光吸收带的吸收)
- 可以通过3级碳自由基的加成可以构筑4级碳中心
- 富电子或者杂环的苄基底物OK
- 杂原子α位碳自由基的手性加成也OK
③ 机理提倡
作为上述的催化循环的一个支撑,作者做了如下工作。
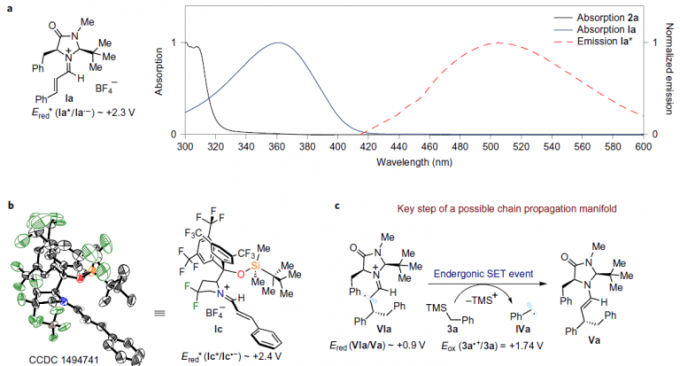
引用自原论文
- MacMillan催化剂与肉桂醛所合成的亚胺正离子,只有在420nm前后有一小段可见光吸收带(上图a)。
- 无光照或者无催化剂存在下反应无法进行。
- TEMPO共存下或者氧气氛围下反应受阻,因此很可能是自由基机理。
- 作者分离出了被激发的亚胺BF4盐的单晶结构。特别是氟取代的吡咯烷结构、观察到了通过gem-氟素导入后引起的特征性配体效应[5](上图b)。
- 亚胺中间体的氧化电位大概在Ered* = +2.3-2.4V(通过CV与荧光波长测定算出)。这是一个可以合理解释催化剂循环的氧化还原热力学的值。
- 苄基TMS以浓度依赖性方式淬灭亚胺阳离子。
- 即使使用Z-肉桂醛进行反应,也可获得与E-肉桂醛相同的立体定向产物。此外,所有回收的原料都变成E体→E / Z亚胺的异构化非常快。
- 没有观察到苄基二聚体的形成、苄基自由基的低亲核性,亚胺离子难猝灭自由基[6]、从苄基TMS到α-亚氨基基团阳离子的SET是吸收性的(上图c)、反应量子收率低(Φ=0.05)等事实来看、自由基连锁机理看来是unlikely pathway。
小编所感
- 光催化剂等效组分并不总是能够独立的持续不断的发挥其功能,常常只有当它与底物结合时才具有可见的光氧化还原能力。因此在设计此类反应的时候,都是考虑以一种组合的方式来达到目的。
- 当然,有机催化剂体系价格低廉,并且由于能够通过共价键固定底物,因此在不对称空间稳健性方面比Ir系统更具吸引力。由于催化剂的结构修饰,拓展的可能性更多,更广泛,因此应用范围似乎很宽。
- 产率总的来说还不错。另外由于2个自由基活性种在附近同时出现,因此更有利于失活防止・产率提高・位阻克服。
未解决的问题
- 该条件对于β-烷基取代的烯酮底物不适用,或许可能通过更短波长的UV光源照射,或者利用其它光催化剂进行能量移动介导等方法实现?
参考文献
- (a) Mariano, P. S. Tetrahedron 1983, 39, 3845. doi:10.1016/S0040-4020(01)90889-0 (b) Mariano, P. S. Acc. Chem. Res. 1983, 16, 130. DOI: 10.1021/ar00088a003
- (a) Arceo, E.; Jurberg, I. D.; Álvarez-Fernández, A.; Melchiorre, P. Nat. Chem.2013, 5, 750. doi:10.1038/nchem.1727 (b) Silvi, M.; Arceo, E.; Jurberg, I. D.; Cassani, C.; Melchiorre, P. J. Am. Chem. Soc. 2015, 137, 6120. DOI: 10.1021/jacs.5b01662 (c) Bahamonde, A.; Melchiorre, P. J. Am. Chem. Soc. 2016, 138, 8019. DOI: 10.1021/jacs.6b04871
- Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265. DOI: 10.1021/cr0680843
- Dockery, K. P. et al. J. Am. Chem. Soc. 1997, 119, 1876. DOI: 10.1021/ja963197x
- Zimmer, L. E.; Sparr, C.; Gilmour, R. Angew. Chem. Int. Ed. 2011, 50, 11860. doi:10.1002/anie.201102027
- Murphy, J. J.; Bastida, D.; Paria, S.; Fagnoni, M.; Melchiorre, P. Nature 2016, 532, 218. doi:10.1038/nature17438
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!














No comments yet.