

本文作者:杉杉
导读
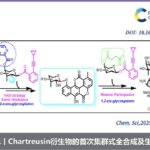
吲哚的氧化重排能够成功构建氧化吲哚(oxindoles)骨架,并且,该反应已经成为构建一系列复杂分子的关键反应。近日,香港科技大学孙建伟与南方科技大学李鹏飞课题组共同合作在Angew. Chem. Int. Ed.中发表论文,报道了通过手性磷酸催化的吲哚的对映选择性氧化重排,进而获得一系列对映体富集的螺环氧化吲哚(spirooxindoles)衍生物。反应过程的对映选择性主要通过动态动力学拆分过程进行控制。
Catalytic Enantioselective Synthesis of Spirooxindoles by Oxidative Rearrangement of Indoles
C. Qian, P. Li, J. Sun, Angew. Chem. Int. Ed. ASAP. DOI:10.1002/anie.202015175.

正文
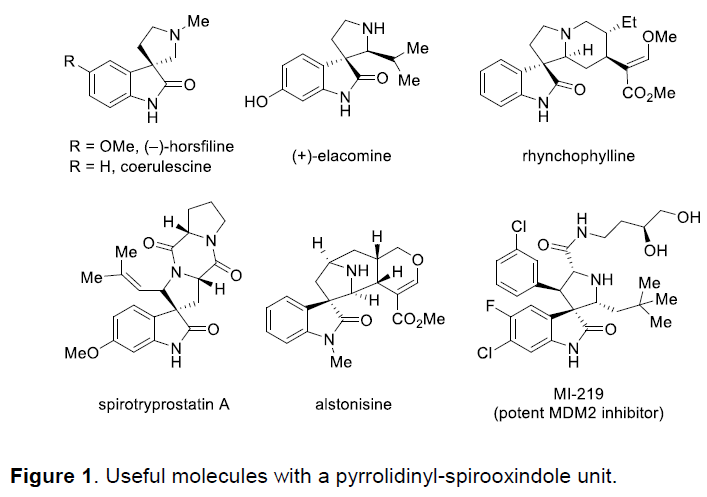
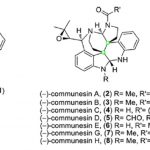
螺环氧化吲哚作为一系列天然产物与生物活性分子中普遍存在的结构单元,尤其是吡咯烷基-螺环氧化吲哚结构单元广泛存在于大量生物碱分子以及一系列治疗药物 (therapeutic agents)分子中(Figure 1)。
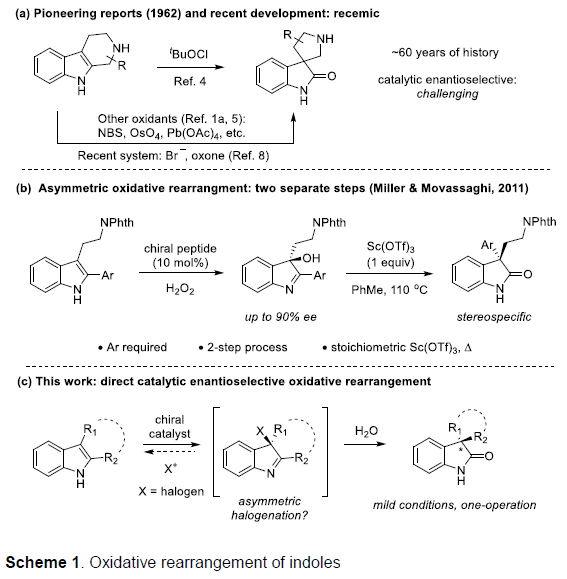
同时,四氢-β-咔啉(carbolines)的仿生氧化重排过程,由于其能够有效地简化合成路线,并且底物易得。因此长期以来备受关注(Scheme 1a)。然而,要实现四氢-β-咔啉的对映选择性仿生氧化重排过程,目前仍然较为困难。2011年,Miller与Movassaghi等[1]巧妙地设计出通过肽催化剂参与的吲哚分子的不对称氧化反应,进而合成出一系列对映体富集的3-羟基假吲哚(3-hydroxyindolenine)化合物,随后,将3-羟基假吲哚通过化学计量的Sc(OTf)3促进的立体专一性重排过程,最终获得一系列对映体富集的氧化吲哚衍生物(Scheme 1b)。然而,由于第二步反应中存在官能团兼容性较差以及反应条件较为苛刻的问题,因此,这种两步反应过程无法通过一锅操作,实现真正意义上的催化反应[2]。而且,吲哚底物中存在的2-芳基取代基同样能够阻碍其在吡咯烷基-螺氧化吲哚合成中的应用。同时,3-hydroxyindolenine重排过程需要较为苛刻的反应条件,可能与3-羟基较弱的离去性能相关。另一方面,亲电卤素源(electrophilic halogen sources)同样能够有效地作为上述重排过程的氧化剂。最近,Tong等[3]报道了一种更加绿色的反应方法学,该方法学中,采用原位生成的Br+作为氧化剂(Scheme 1a)。受上述研究的启发,作者假设将吲哚3-位进行催化不对称卤化反应,可能会获得对映体富集的3-卤代假吲哚(3-halo-indolenine)中间体,同时借助卤离子良好的离去性能,从而使后续的立体专一性重排反应能够在温和的条件下进行(Scheme 1c)。
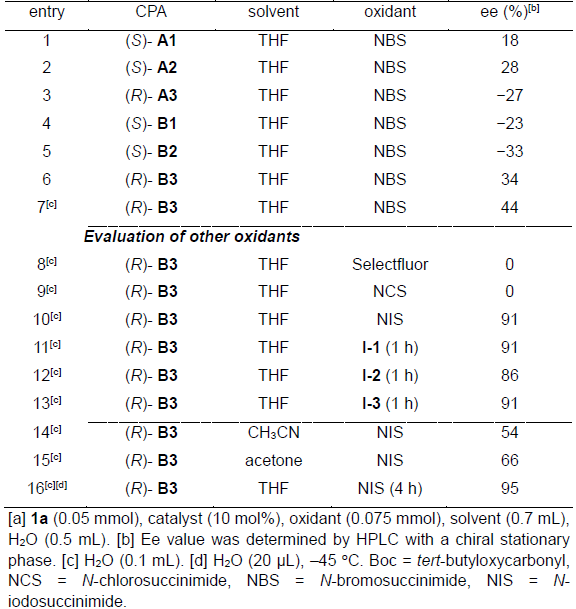
首先,作者以N-Boc保护的四氢-β-咔啉1a作为底物,分别对手性磷酸 (chiral phosphoric acid, CPA)催化剂、氧化剂、溶剂等进行相应的优化筛选(Table 1)。最终确定最佳的反应条件为,采用10 mol%的(R)-B3为手性磷酸催化剂,1.5当量的NIS作为氧化剂,在THF/H2O(v/v = 35:1)的混合溶剂中,−45℃下进行反应,最终获得95%ee的目标产物2a。
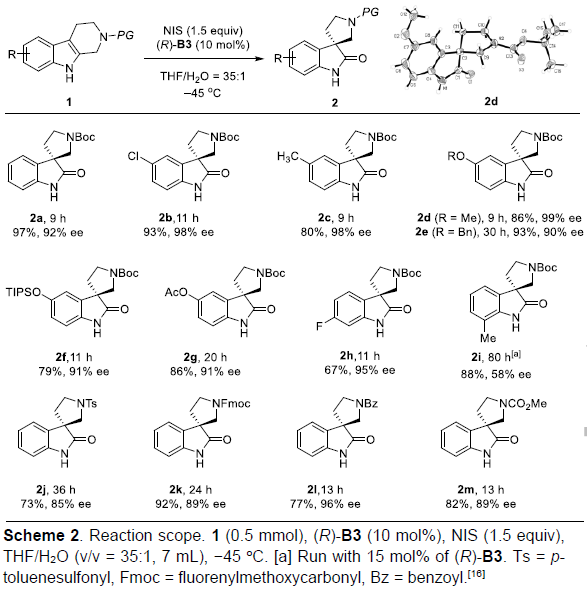
在获得上述最佳条件之后,作者首先考察四氢-β-咔啉底物1的应用范围(Scheme 2)。实验结果表明,上述反应条件对于四氢-β-咔啉环中的各种不同类型的取代基均能够良好地兼容,进而获得相应的吡咯烷基-螺环氧化吲哚产物2a–2m。而对于2i,可能存在邻近的甲基取代基,从而阻碍N-1位氢键受体与CPA之间形成有效的氢键相互作用,因此,2i在上述条件下具有较低的反应性与较差的对映选择性。
随后,该小组通过对上述条件的再次优化,最终发现,选用(R)-A4为催化剂时,上述反应还能够顺利完成一系列对映体富集的四氢呋喃基-螺环氧化吲哚分子4a–4f的构建(Scheme 3)。值得注意的是,这类化合物较易结晶,因此,能够通过结晶的方法,进一步提高其对映纯度。
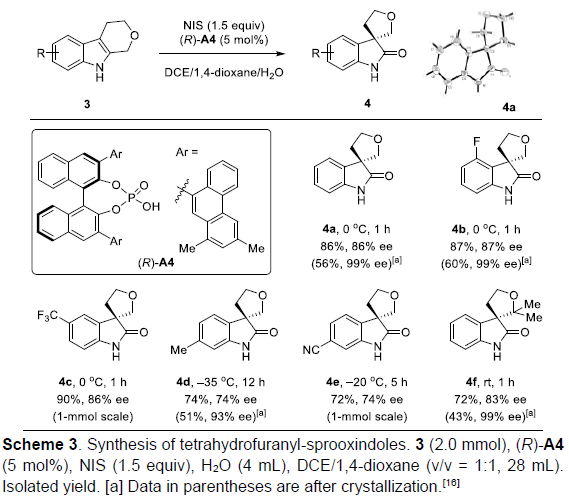
同时,作者对其它不同类型的底物进行进一步研究(Scheme 4)。例如,吲哚1o在(R)-B2催化剂存在下,可以获得高度对映体富集的3,3-二取代氧化吲哚2o。而吲哚5a–5b在(R)-A4催化剂存在下,最终获得在2-位具有环稠合的产物6a–6b。尽管这种反向重排 (inverted rearrangement)已有文献报道[4],然而,有趣的是这种底物中取代基的细微差异,却能够产生明显不同的区域选择性控制。已有研究表明,杂原子甲基取代基具有更高的迁移趋势[5]。然而,对于含有七元含氮杂环稠合的吲哚1n底物而言,并未观察到重排过程的发生。
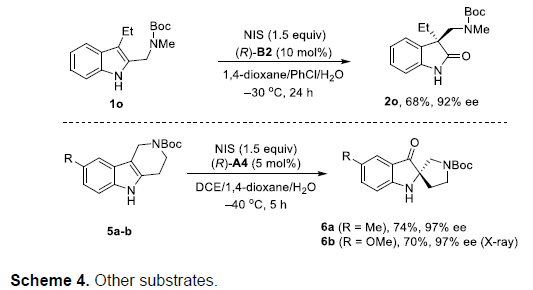
接下来,作者进一步发现,将1mmol 1的d在同样条件下反应,获得的(R)-2d产物通过后续几步反应,进而顺利完成天然产物(-)-horsfilin的全合成。与之前的文献报道相比[6],这一策略更加有效,并且无需选择手性辅基(Scheme 5)。
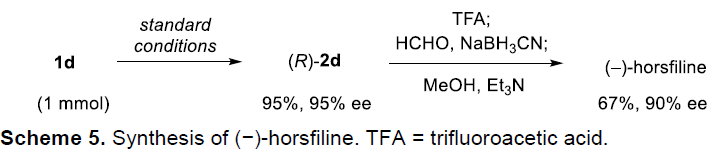
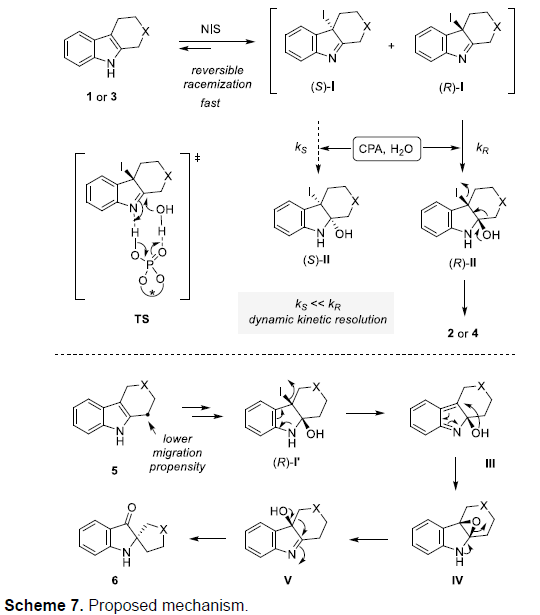
为了进一步理解上述反应的机理,作者进行一系列相应的控制实验研究(Scheme 6)。首先,将1a在标准条件下进行上述转化,并通过NMR,观察可中间体IM的形成,而IM中间体十分不稳定,无法分离获取(Scheme 6a)。然而,令人欣喜的是,选择底物3f进行上述转化时,却能够获得足够稳定的外消旋形式的中间体7f,并成功进行分离与结构表征。然而,在选用(R)-A4催化剂参与氧化重排时,能够获得86%ee的氧化吲哚产物4f。上述实验结果能够排除不对称卤化过程作为对映体决定步骤的机理路径(Scheme 6b)。同时,作者观察到7f溶液在反应过程中逐渐变为紫色(可能是由于碘的形成),同时观察到3f底物的生成。这一现象表明卤化过程是可逆的。随后,作者进一步将3a与7f混合,在A4催化剂存在以及未加入NIS的条件下,进行交叉实验,最终获得60%收率与87%ee的交叉反应产物4a,同时,7f完全转化为3f。这些结果进一步证实了卤化步骤是可逆的(Scheme 6c),而后续的反应步骤决定整个反应过程的对映选择性。
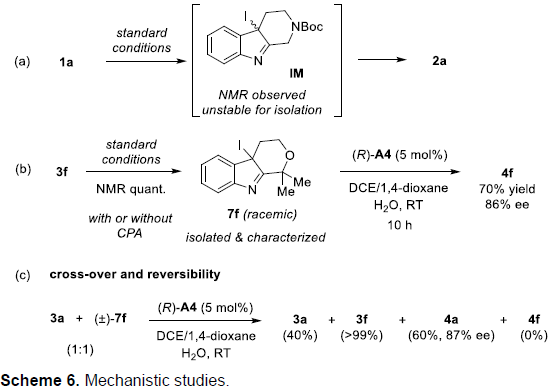
基于上述的实验观察,作者提出了可能的反应机理(Scheme 7)首先,在吲哚3-位迅速并可逆地进行卤化过程,形成外消旋中间体I。随后,I在CPA作用下,形成活化的亚胺中间体,并进一步与水加成,通过过渡态TS,获得缩醛胺(hemiaminal)中间体II。在过渡态TS中,CPA作为双功能催化剂 (bifunctional catalyst),同时活化亚胺I与水。因此,I的两种对映中间体通过手性催化剂CPA的手性识别作用,能够以不同的速率参与上述反应,其中,(R)-I能够更加迅速地完成后续反应。而反应速率较慢的(S)-I中间体,通过可逆的卤化作用发生外消旋化,从而有效地实现相应的动态动力学拆分 (DKR)过程。最后,通过(R)-I形成的(R)-II异构体,进一步经过1,2-迁移,最终获得对映体富集的氧化吲哚产物。此外,为进一步解释底物5(3-位取代基具有更高迁移趋势)的反向重排,作者假设获得(R)-I’反应步骤均与获得(R)-I相同。(R)-I’进一步形成中间体III。然而,由于中间体III中2-取代基的迁移性能较差,因此,中间体III可能经过环化过程,形成环氧化物IV。最后,IV在胺氮原子中孤对电子的协助下,开环形成中间体V,再进一步通过半频哪醇类型重排,获得产物6。
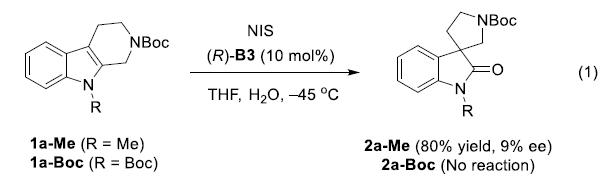
此外,作者还对N-保护的底物1a-Me与1a-Boc的反应进行进一步研究(eq 1),结果表明,1a-Me底物能够获得相应氧化吲哚产物2a-Me,然而,无法获得优良的对映选择性,这表明氢键对于反应过程中对映选择性的控制至关重要。而1a-Boc则无法有效地完成上述转化,可能源于Boc基团的存在,使吲哚环的电子密度显著降低。
总结
香港科技大学孙建伟与南方科技大学李鹏飞课题组共通合作报道了采用NIS作为氧化剂,CPA作为手性催化剂,在温和的反应条件下,通过吲哚的氧化重排方法学,顺利获得一系列对映体富集的螺环氧化吲哚衍生物。该方法学具有较高的产物收率与优良的对映选择性。同时,作者进一步将该方法学应用于 (-)-horsfiline的全合成。上述转化过程的反应机理涉及迅速可逆的卤化过程、水加成以及重排过程。并且,外消旋有机卤中间体的动态动力学拆分,实现上述反应过程的对映选择性的控制。













No comments yet.