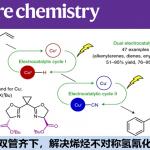
C-H的直接官能团化一直是近几年的研究热点。特别是利用一些过渡金属进行的选择性C-H活化不管是作为方法学,还是在全合成应用中都是竞相追逐的潮流之一了。
由于小编是做催化剂设计并且应用于选择性C-H活化研究的,所以在以往的记事中也都偏重于这一块(有兴趣的可以回顾一下)。
在实现区域选择性C-H活化上,现在主要还是依靠DG(导向基)的作用,虽然这种方法非常实用,近年来也有一堆好文刊出。但是从小编的视角来看,这种只能算作是底物控制的选择性反应,与利用催化剂控制(氢键相互作用等非立体位阻效应)产生的选择性方法相比还是差了一个档次,这也是利用催化剂准确命中芳香环间位这篇文章,虽然反应本身并不新颖,但是还是能够上nat.chem的原因了。
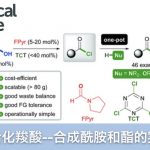
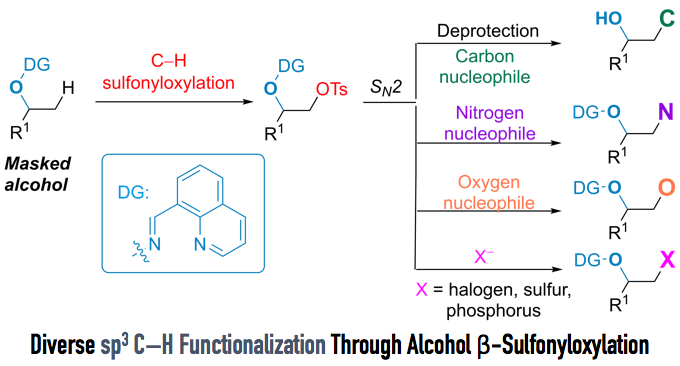
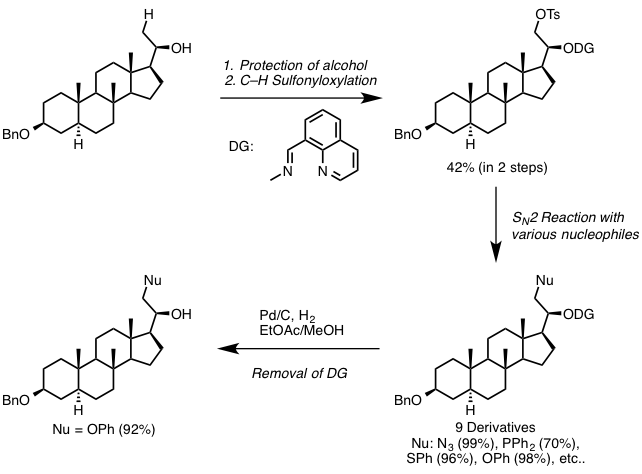
而今天要介绍的一篇文章,同样是利用DG进行的β位的sp3 C–H的OTs化反应,然后紧接着利用SN2反应进行的各种后续官能团化。
该方法由德州奥斯汀分校的董广彬教授(化学家专访3月期)近期发表在nat.chem上。读完这篇文章只能感慨,董老师的strategy每次都是这么好,仅仅只是方法改进的一小步,可是在实用性上确实一大步。今天小编想在这里介绍一下这篇文章
Xu, Y.; Yan, G. B.; Ren, Z.; Dong, G. B. “Diverse sp3 C−H Functionalization Through Alcohol β-Sulfonyloxylation”. Nature Chem.2015, 7, 829-834. DOI: 10.1038/nchem.2326
背景介绍-直接SP3C-H键的官能团化的典型实例
A) Rh-催化的分子内C-H胺基化(Du bios, J. Am. Chem. Soc. 2001, 123, 6935.)
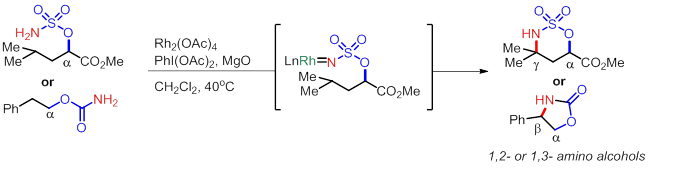 B) 类Hofmann-Loffler-Freytag自由基反应 (Phil S. Baran, J. Am. Chem. Soc. 2008, 130, 7247–7249.)
B) 类Hofmann-Loffler-Freytag自由基反应 (Phil S. Baran, J. Am. Chem. Soc. 2008, 130, 7247–7249.)
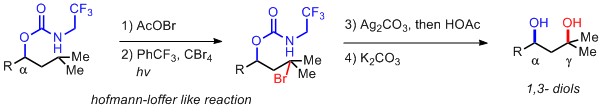 C) Ir-催化脱氢C-Si化反应(John F. Hartwig, Nature 2012, 483, 70.)
C) Ir-催化脱氢C-Si化反应(John F. Hartwig, Nature 2012, 483, 70.)
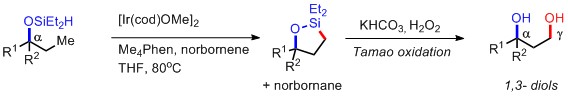 D) 醇β位的催化SP3C-H的OAc化(GB. Dong. J. Am. Chem. Soc. 2012, 134, 16991)
D) 醇β位的催化SP3C-H的OAc化(GB. Dong. J. Am. Chem. Soc. 2012, 134, 16991)
由于羟基或者氨基普遍存在于很多活性小分子中,同时这两个明星基团又可以转化成各种其他官能团,所以在在1,2-, 1,3-官能团化尤其是形成diol或者amino alcohol的反应开发上,近几年可谓是成果显著。但是,以上的A),B) ,C)三个反应中有一个比较大的限制就是无法形成化学相异的羟基,所以在进一步的转化中选择性就成了一个问题。而在实例 D) 中,Dong的方法就有所突破,最终得到的是O-DG与OAc两个不同保护基保护的羟基,虽然仅仅是这一个看似不大的变化,但是很简单的就把两个羟基区分开,实用性大大改善。
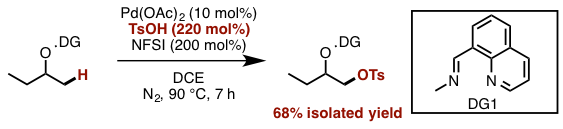
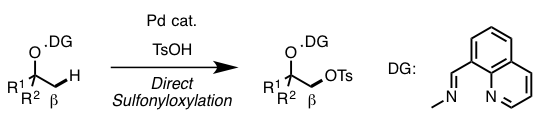
在今天要介绍的反应中(上图),Dong在筛选了很多DG后,最终发现利用DG1并且直接加入2.2当量的TsOH可以有效地对β位的sp3 C–H进行-OTs化反应,由于-OTs的邻位是饱和的CH2所以反应并没有造成-OTs的消除离去,最终形成了如图所示的1,2-chemical differentiated diols(68% isolated yield)。并且该反应在水与敞开环境下都能够稳定进行。
本反应的机理解析探索(纯属小编个人看法)
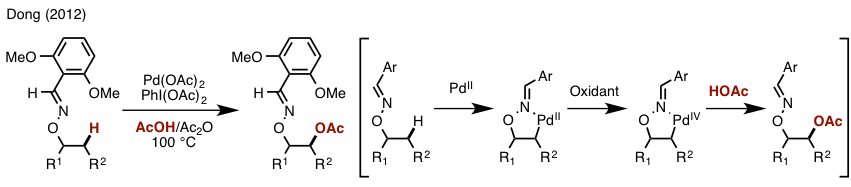
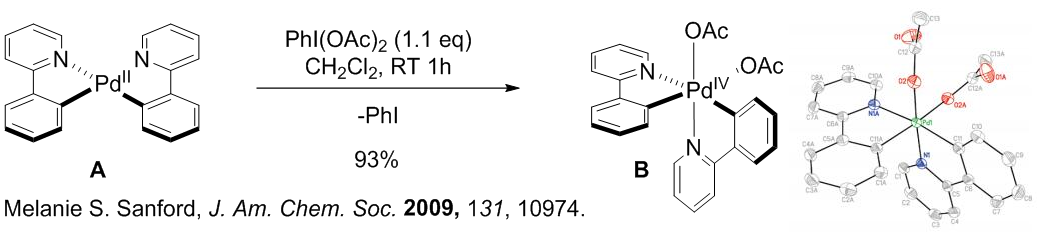
该反应中我们可以看到使用了氧化剂NFSI(亲电氟化试剂),在催化循环中NFSI是作为亲电的氧化剂把Pd(II)氧化成Pd(IV)催化整个反应的。那么小编也找到了这类亲电氧化剂作用的证据。下图是sanford组利用高价碘试剂与Pd(II) complex在室温下反应得到的氧化后的Pd(IV) complex,其结构也通过X-ray解析验证。而本文中用到的NFSI其实是具有异曲同工的作用,由于氧化后的F配位的产物结晶比较困难,所以小编在这边也不能给出一目了然的实例。
在弄明白NFSI的作用后,我们还剩下一个很大的疑问,就是在reductive elimination阶段,由于Pd(IV)上可能会连有-OAc, -OTs, -F, -N(SO2Ph)2,那么到底是一个怎样的选择性还原消除过程呢。为了弄明白这个,小编可谓是查了不下三十篇文献。最终皇天不负有心人啊,终于找到了下图所示的可谓是证据吧 (Guosheng Liu, Chem. Sci., 2013, 4, 3172.)。
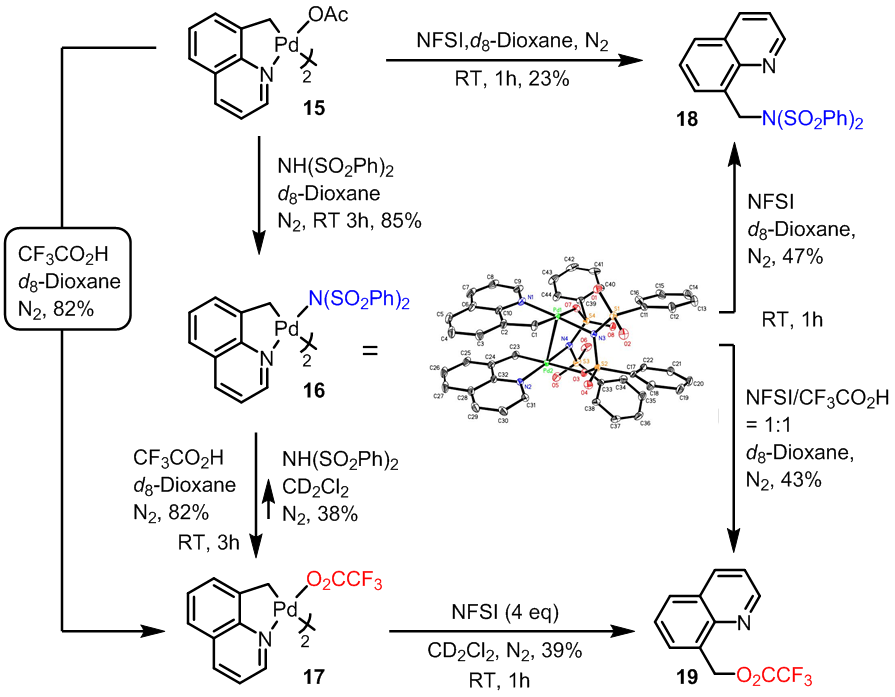
从上图可以看出,OAc配位的Pd(II) complex 15在室温下可以很容易的发生ligand exchange形成 -N(SO2Ph)2配位的16, 或者三氟乙酸配位的complex 17,而在16与17之间的变换反应中我们可以发现CF3CO2H可以很容易的与-N(SO2Ph)2发生置换把16变成17,而从17变成16的话,从低产率可以看出相对来说困难很多。而16在加入氧化剂NFSI后可以得到C-N偶联的产物,但是另一方面在加入同等当量CF3CO2H的条件下,由于ligand exchange的作用,得到的确是C-O偶联的产物19, 同时在17中直接加入NFSI也是选择性的得到C-O偶联的产物19,从这一张图中我们可以看出,CF3CO2–,(PhSO2)2N–,CH3CO2–,这三个配体,它们的还原消除顺序应该是CF3CO2–>(PhSO2)2N–>CH3CO2–,与他们的酸性成正比(强酸制弱酸),与亲核性成反比。
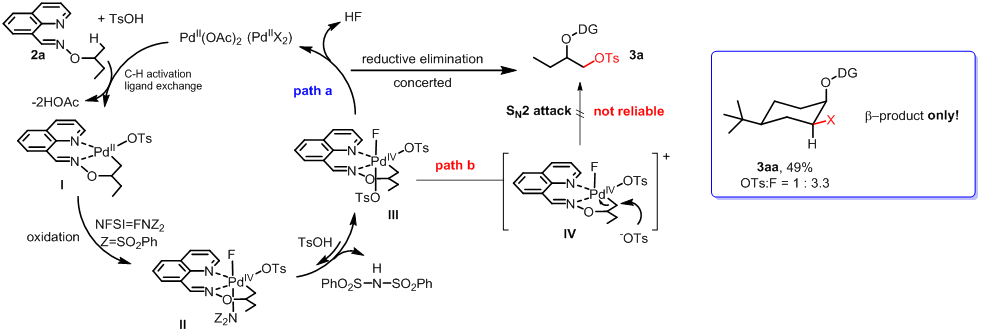
所以根据以上的文献调查,或者也可以说是启发吧,小编斗胆自己推测了反应机理(上图),首先从Pd(OAc)2出发,通过螯合配位辅助的C-H活化并且其中伴随着TsO-与OAc的ligand exchange形成中间体I, 然后在被NFSI氧化形成Pd(IV)中间体II,在这一步再次进行ligand exchange脱去NH(SO2Ph)2后形成中间体III,在这里有两种可能性,一种就是concerted reductive elimination(path a), 而另一种是通过OTs的SN2亲核进攻进行的reductive elimination(path b).而在一些亚甲基的C-H活化产物中小编发现一个规律,就得到的产物如3aa那样的β-位的单非対映异构体,这间接地说明至少对于亚甲基底物的C-H活化反应path b并不存在,另外-OTs的亲核性也很弱,这也必须考虑在内。
本反应的应用
利用该反应得到-OTs取代产物后,继而可以利用SN2反应生成各种C-C,C-N,C-O,以及很难通过C-H直接活化得到的C-S,C-P键产物。本文中利用steroid作为底物,进行了很多后续的修饰,引入一些活性小分子等等,验证了该反应的实用性。并且DG可以选择性的在温和条件下(Pd/C)脱除,意义巨大。
小编所感
就像小编在开头所说,其实这种直接的C-O生成反应早在2000年初就已经被发现。而董广彬教授总是能够从实用性出发,在别人的基础上更进一步,他的一些方法学设计经常都是画龙点睛之笔,不得不赞。而对于C-O生成的机理研究到目前为止还没有一个广泛接受的定论,文章仅仅是小编个人的一些浅薄的看法。高价金属的热度最近有逐渐上升的趋势,对于四价Pd来说,就像本文中的底物实例(详见论文原文),由于四价Pd complex的刚性以及立体位阻比较大,虽然这降低了beta-消除的副反应几率,但是也使得反应只能针对一些立体位阻小的比如甲基的C-H活化,正可谓有利也有弊吧,但是其很高的反应性也燃起了C-H活化的第二春。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!