
作者:石油醚
导读:
近日,浙江工业大学叶欣艺研究员、王鸿教授联合南洋理工大学陈俊丰(Tan Choon-Hong)教授以及澳大利亚伍龙贡大学Richmond Lee教授利用离子对催化策略开发了一种高效的叔胺不对称氧化方法。在这项工作中,作者探索了环状和非环状叔胺,在大多数情况下该反应都表现出显著的对映选择性,并且底物无需使用大位阻取代基。
“Asymmetric N-oxidation Catalyzed by Bisguanidinium Dinuclear Oxodiperoxomolybdosulfate.
Wentao Wu, Esther Cai Xia Ang, Xinru Xu, Qi Wang, Hong Wang*, Richmond Lee*, Choon-Hong Tan* and Xinyi Ye*
Nat Commun, 2024, 15, 7317. doi: 10.1038/s41467-024-51765-0”
正文:
手性氮氧化合物广泛存在于在众多天然产物和生物活性物质当中,包括乙酰胆碱酯酶抑制剂,腺苷摄取抑制剂,抗肿瘤药,NF-kB抑制剂以及MAO-B抑制剂等(图1a);此外,手性氮氧化合物也表现了卓越的配体特性,在有机金属化学中发挥着重要的作用(图1b)。直接氧化叔胺是获得手性氮氧化物的最简洁高效途径。然而由于氮原子上的孤对电子会快速翻转,导致氧化过程手性难以控制,以高对映选择性得到氮氧化物仍然是个巨大的挑战。Colonna和同事们最初发现了牛血清白蛋白(BSA)介导不对称硫氧化的催化潜力,随后Gaggero小组推进了BSA催化的氧化体系,将其应用扩展到氮氧化上,但难以实现较高的对映选择性。Ottolina和同事们分离了环己酮单加氧酶(CYMO),利用辅因子NADPH和氧气实现了叔胺的不对称氧化,后来Tang团队在此基础上开发了一类基于四氧嘧啶骨架的手性催化剂,将对映选择性提高到了93%,但该反应对底物有极大的限制(图1c)。在2016年,Yamamoto团队设计了一种具有联二萘酚骨架的双金属中心催化剂,也实现了叔胺的不对称氧化(图1d),不过催化效果仍然是差强人意。以上这些策略说明要想扩大叔胺氧化的底物范围和实现高对映选择性,需要更强大的诱导效应或空间位阻来阻止叔胺氮上孤对电子的快速消旋。
近日,浙江工业大学叶欣艺研究员、王鸿教授联合南洋理工大学陈俊丰(Tan Choon-Hong)教授以及澳大利亚伍龙贡大学Richmond Lee教授利用离子对催化策略开发了一种高效的叔胺不对称氧化方法。在这项工作中,作者探索了环状和非环状叔胺,在大多数情况下该反应都表现出显著的对映选择性,并且底物无需使用大位阻取代基(图1d)。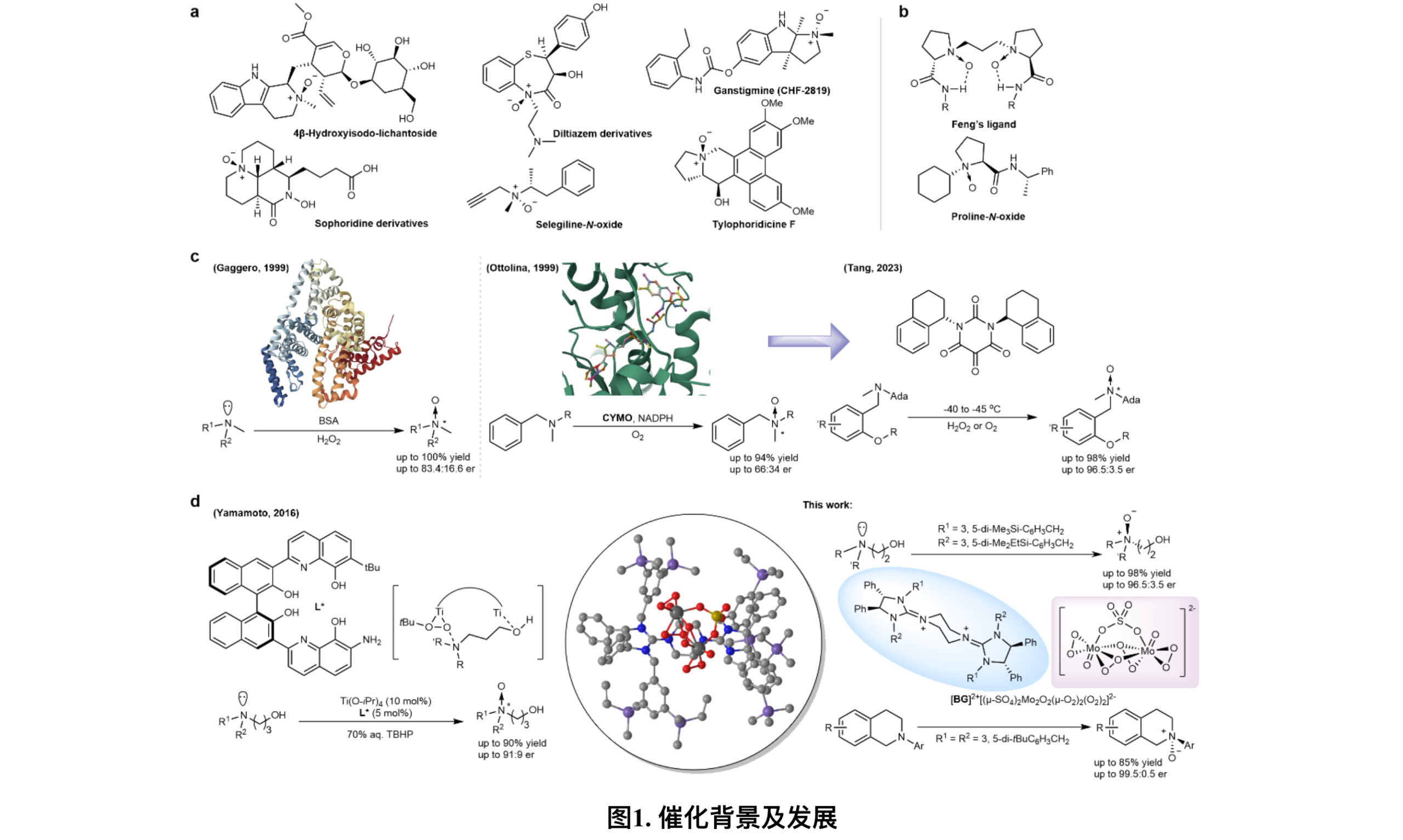
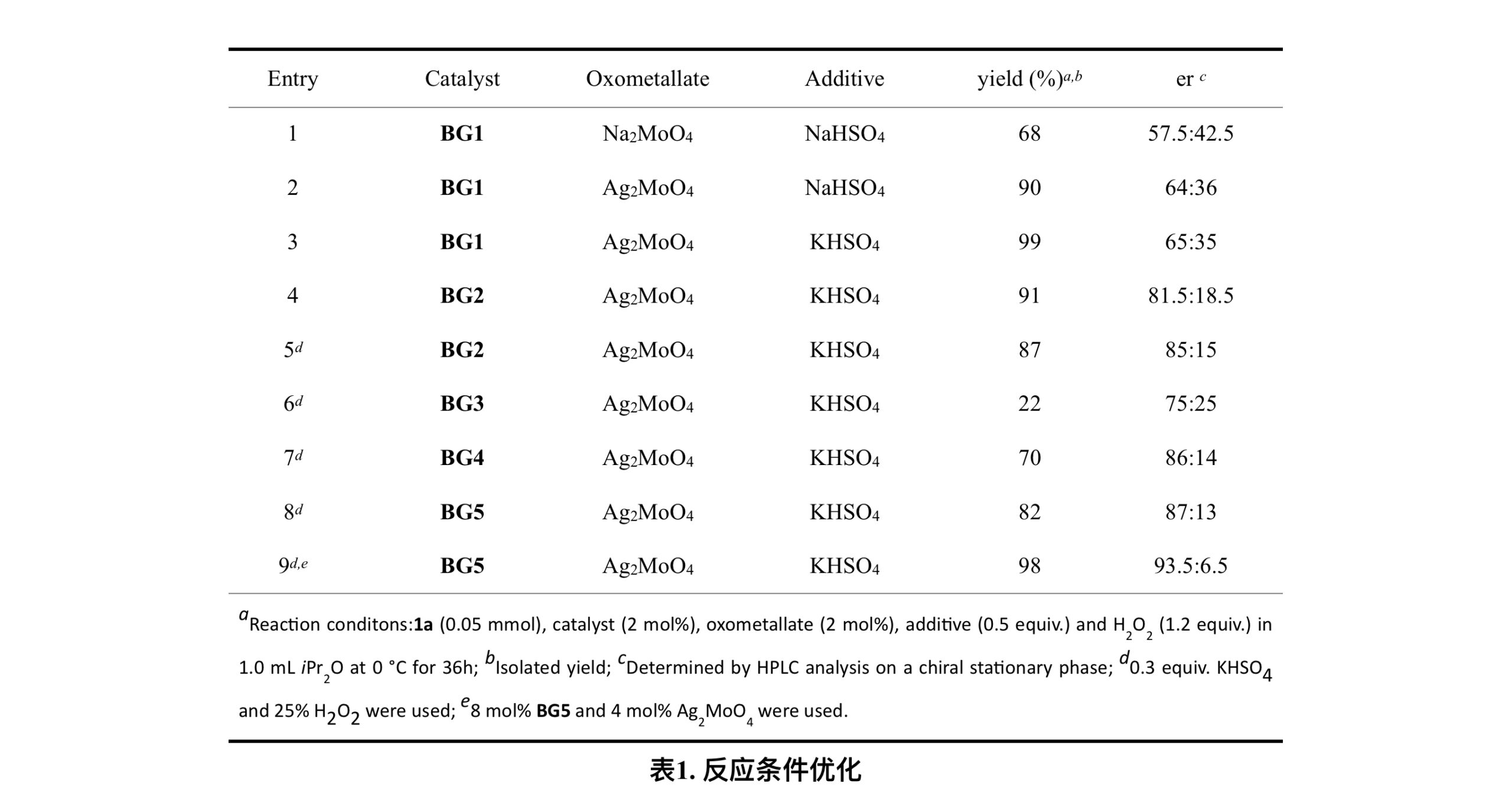
在工作初始阶段,作者尝试了各种双胍阳离子催化的氧化体系来评估它们对氮氧化的适用性。但不幸的是,像高锰酸盐和钨酸盐这样的强氧化剂会将苄位的亚甲基氧化从而形成酰胺,而钼酸盐则能够以高化学选择性来氧化叔胺氮。通过添加剂筛选和催化剂改造,最终他们创造性地在BG的四个侧链的正交空间内同时放置TMS和DMES,得到的BG5能够以98%的产率和87%的ee实现叔胺的不对称氧化(entries 8-9)。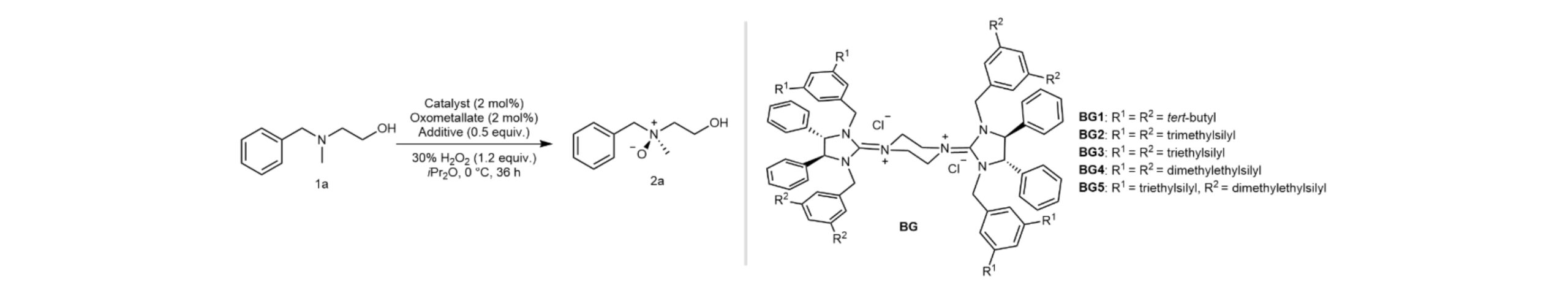
有了优化的反应条件,作者继续探索了烷基胺不对称氮氧化的底物范围(图2)。在苄基苯环对位上的取代,无论是吸电子取代基(2b-2i)还是给电子取代基(2j-2n)反应都能很好地耐受,以高产率(80-98%)和高对映体比率(er)(90:10至96.5:3.5)得到对应手性氮氧化物。该反应对与其他芳环也有很好的兼容性(2x-2z),但1-萘基(2y)对催化剂的手性识别有一定的阻碍作用。为了研究底物上的β-羟基作为导向基团的作用,作者分别在苯基邻位(1aa)和氮γ位(1ab)引入了羟基。2aa可以92:8 er获得,表明手性识别未受影响,但在2ab只以低产率生成了消旋氮氧化物;然而当羟基被甲基(2ae)替换时,产率和ee有了显著的下降。这意味着羟基可能发挥了导向功能,并且其与氮中心之间的距离具有关键的作用。进一步的底物修饰表明当胺上的甲基被乙基(2ac)替换时,不对称氧化受到阻碍(与2a相比),而当增加苯基和氮原子之间的碳链长度时,叔胺则难以被氧化。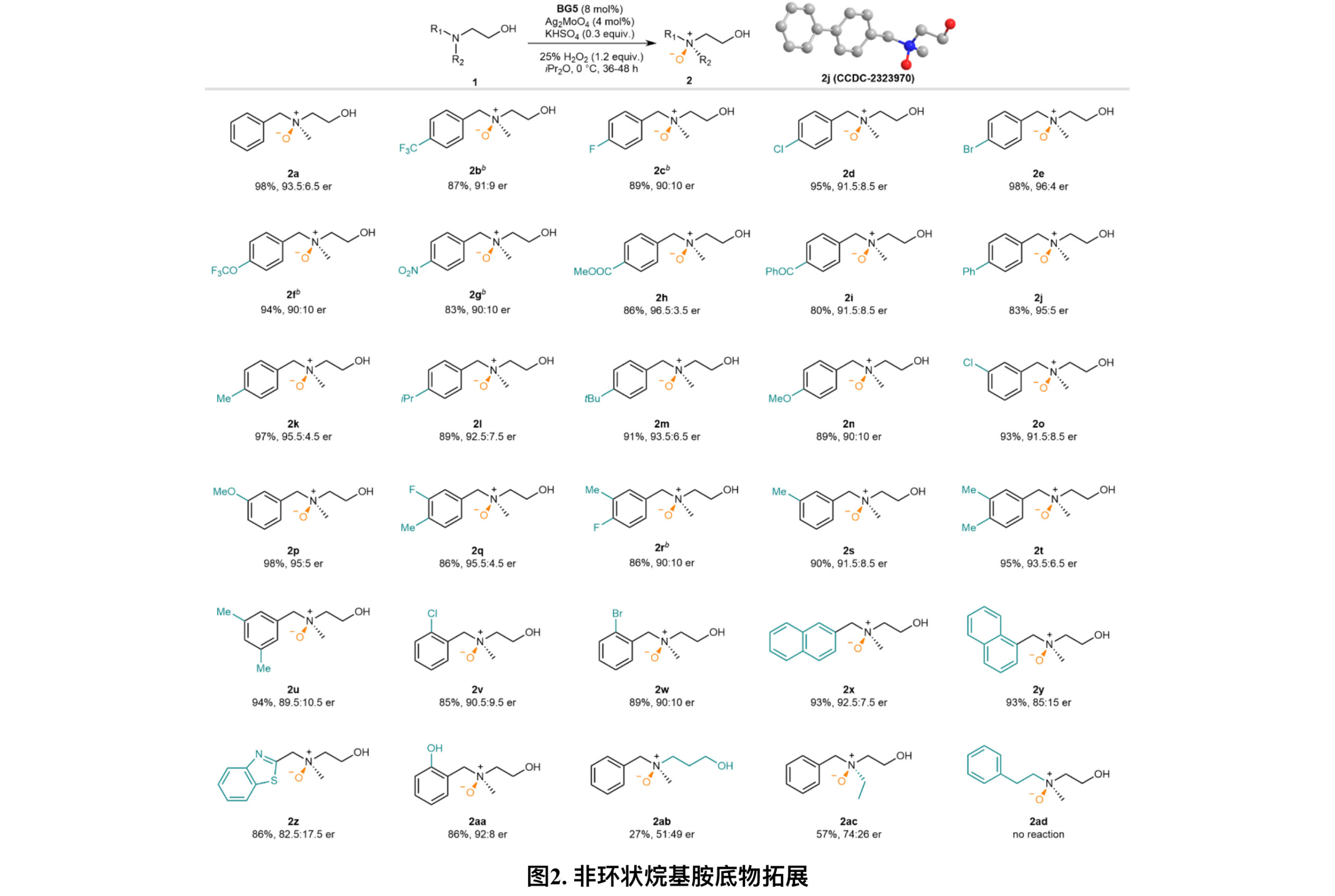
与烷基胺不同的是,芳胺中氮的孤对电子由于p-π共轭效应,更加难以实现不对称氧化,到目前为止还未有报道实现该反应的方法(图3在筛选了催化剂侧链上各种较小的取代基后,作者发现BG1产生了更好的结果,并之后进行了系统的反应条件优化,能以70%的产率和96:4 er获得对应的叔胺氧化物4a。其他四氢异喹啉的底物在新反应条件下也都表现出令人满意的反应性,以中等产率和优异的对映选择性生成了4b-4g。值得注意的是,氮中心周围的空间位阻有利于手性诱导(4h-4j)。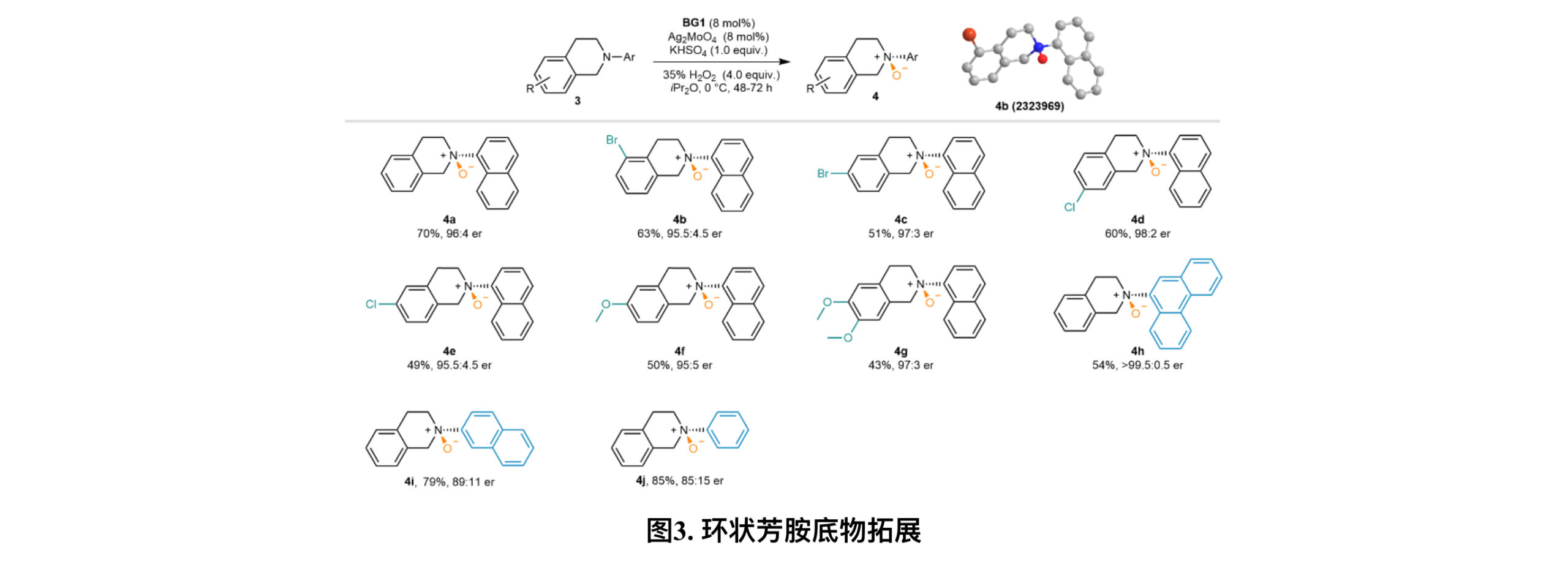
为了展示该不对称氧化的实用性,作者首先进行了克级反应,实现了高产率和良好的对映选择性。在手性氮氧化物的烷基链上存在末端羟基的情况下,通过直接酯化,可以轻松引入某些药物分子(图4)。通过不对称氧化策略也能够直接合成抗组胺药曲美他嗪的类似物13,表明了该方法在获取一系列药物类似物方面的多样性和适用性。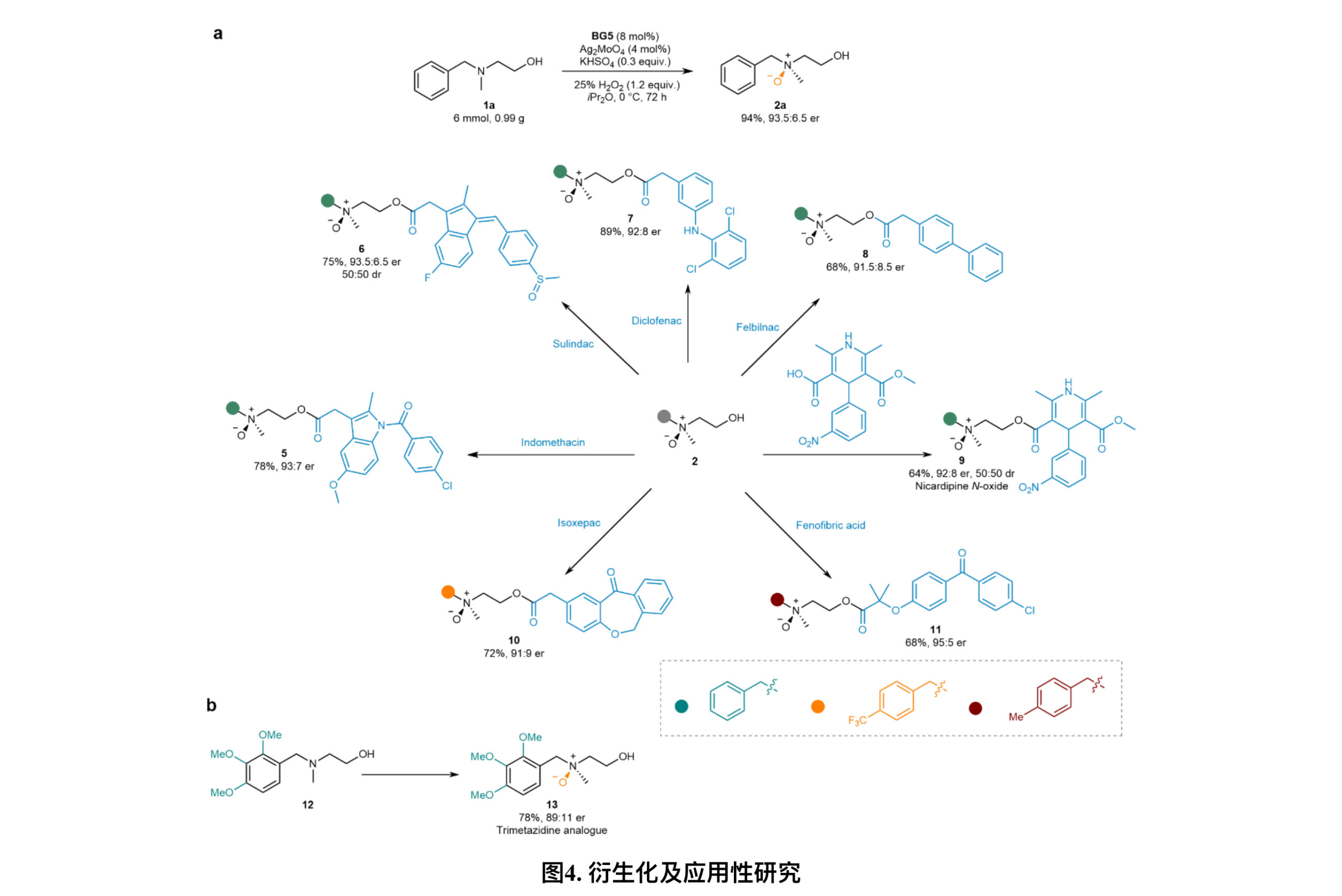
为了寻找到实际发挥氧化作用的催化剂,作者在模拟反应条件但不添加底物的情况下得到了BG6,并且1a可以被BG6直接氧化,无需额外的氧化剂。当BG6的负载量分别为0.1和0.25当量时,能以8%和24%产率生成2a(图5b),说明杂多酸阴离子中两个端位过氧基团只有一个参与了氧化过程。为了进一步了解反应机理,作者随后也开展了动力学实验。在标准条件下,最初6小时内产物缓慢累积(图5d),而使用BG6作为催化剂时产率随时间线性增加(图5e,5f),但在所有动力学实验中,2a的ee值都逐渐增加直到反应中期,说明氧化过程中还存在其他反应。因此作者猜测氮氧化物上的氧可能会重新氧化底物,从而导致产物消旋。为了证实该消旋化过程,作者后续向2a的异丙醇溶液中加入1.0当量的1a,ee值从87%下降到78%。此外,BG6和2a的ee值表现出了出线性关系,说明活性催化剂BG6在很大程度上主导了反应的对映选择性(图5h)。Hammett图展现了良好的线性关系(除氟取代基),且为负斜率(ρ = -0.5116),证明了反应过程中氮上会有正电荷累积的现象。对于对氟取代基团的偏差,通过19氟谱分析可以检测到(图5j),因此作者猜测还原状态下的催化剂与氟基团仍然有一定的相互作用。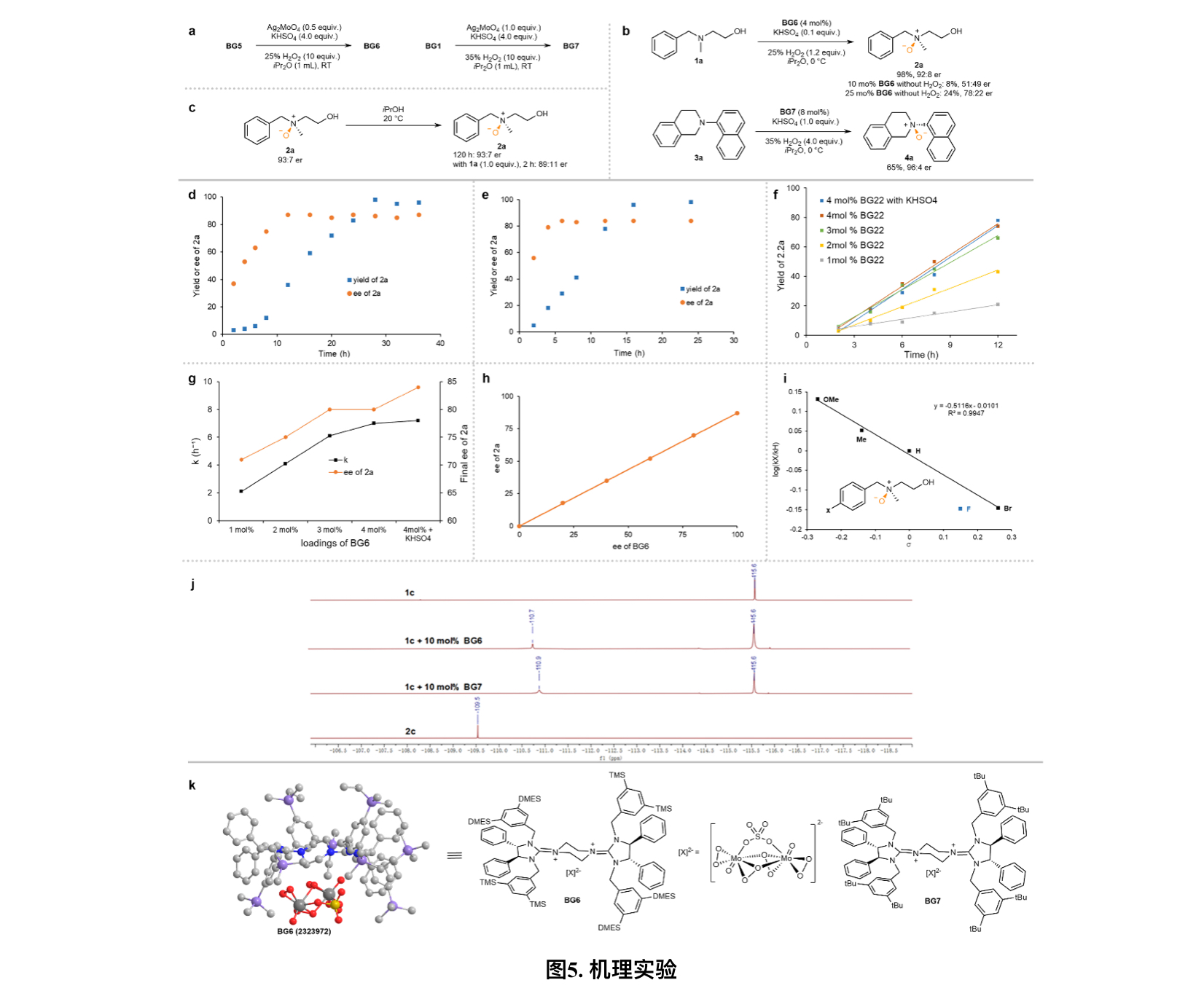
最后作者也使用密度泛函理论(DFT)计算并模拟了催化剂BG6与1a之间的相互作用,生成了R-或S-构型氮氧化物的过渡态结构。TSC_S是最稳定的(图6),与BG6和1a的相对能垒ΔG‡sol为19.0 kcal/mol,其次是TSA_R,ΔG‡sol为20.6 kcal/mol。DFT模型预测(S)-氮氧化物将是主要的对映体,并且通过比较TSA_R和TSC_S,理论e.r.计算为95,ΔΔG‡sol为1.6 kcal/mol,与2a的实验e.r.(94)相吻合。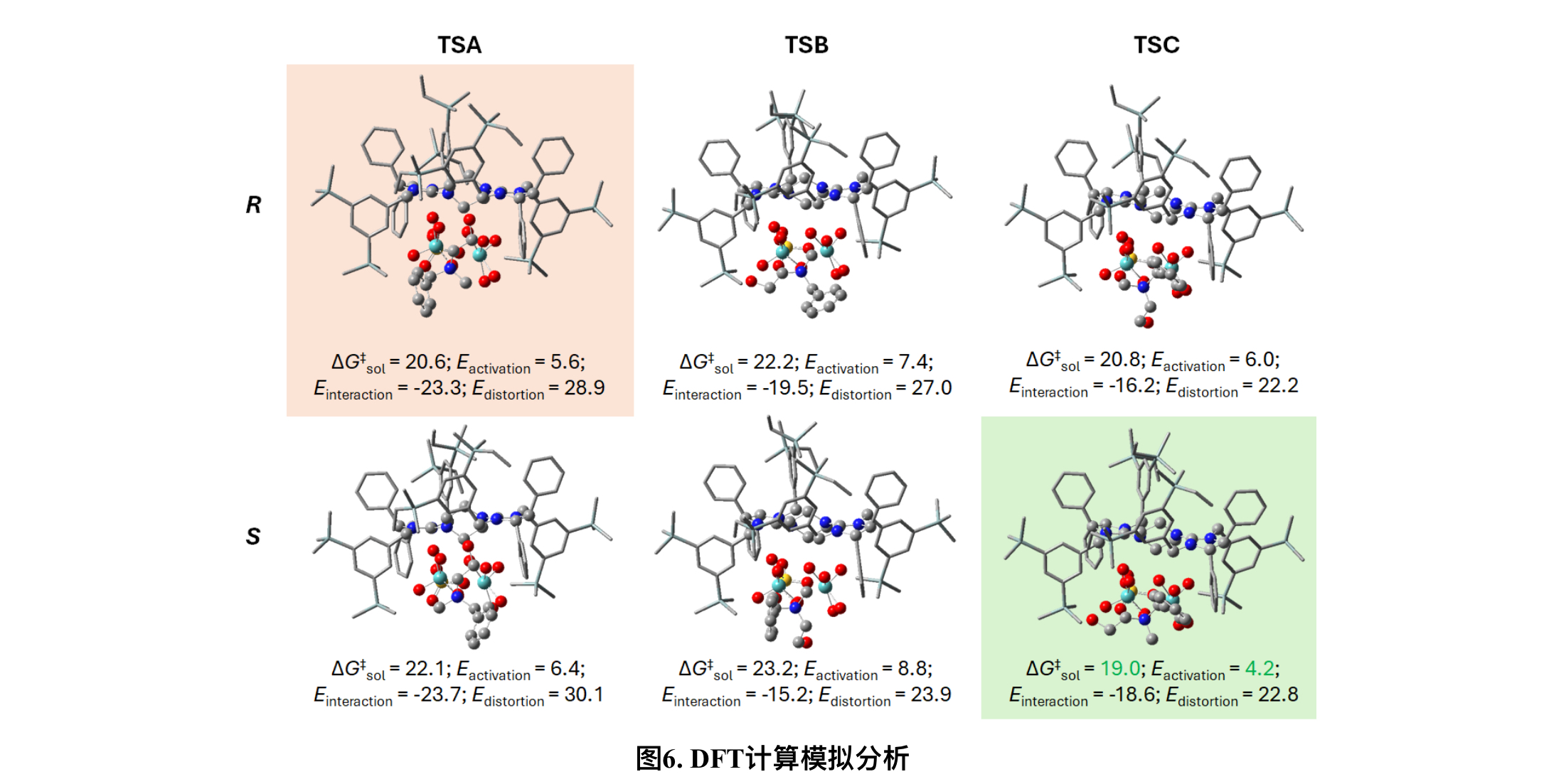
基于以上结果,作者提出了一个合理的催化循环(图7):在水相中生成活性阴离子物种A,随后A被手性阳离子BG2+捕获并转移到有机相中,生成氧化性离子对B。接着1a与B的一个侧边过氧基团发生作用,发生分子间的氧转移从而产生手性氮氧化物(S)-2a,B则恢复到其还原状态C。最后,C的阴离子部分转回到水相中并再次被氧化,从而完成整个催化循环。同时,(S)-2a上的氧原子表现出了一定的氧化能力,以相对较慢的速率氧化胺底物1a产生(rac)-2a。这个过程可以被1a或2a的质子化所阻碍。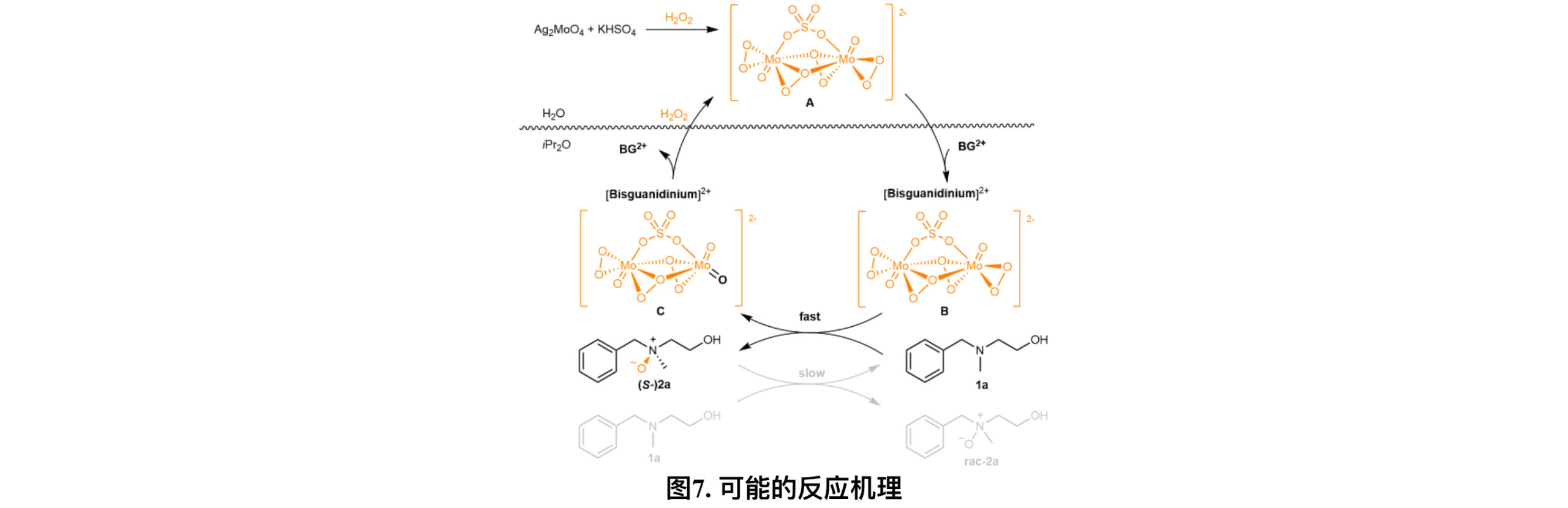
该工作近日发表在Nature旗下Nature Communications期刊上,浙江工业大学为第一通讯单位,澳大利亚卧龙岗大学,南洋理工大学的多位研究人员参与了研究。吴文韬博士为文章的第一作者,王鸿教授、Richmond Lee教授、陈俊丰(Tan Choon-Hong)教授以及叶欣艺研究员为该文章的共同通讯作者。
(非常感谢浙江工业大学王鸿/叶欣艺教授对化学空间的支持)
王鸿/叶欣艺教授课题组简介:

叶欣艺,运河青年学者,硕士生导师,主要从事不对称离子对催化方向的研究,主持国家重点研发计划课题,国家自然科学基金青年基金等项目。迄今以第一/通讯作者Nat Commun.、Angew. Chem. Int. Ed.、ACS Catal.、Chem. Sci.、Chem. Catal. Org. Lett.等多个国际期刊上发表论文17篇。
王鸿,浙江工业大学健行学院执行院长,药学院、绿色制药协同创新中心教授,博士生导师,海洋药物团队负责人,浙江省“万人计划”科技创新领军人才。现任中国药学会海洋药物专委会委员、中国药理学会海洋药物药理专委会委员、中国海洋湖沼学会药学分会理事、浙江省高等学校药学和中药学类专业本科教学指导委员会委员等职。现为海洋药物领域国际权威学术期刊Mar.Drugs编委及二十余个国际学术期刊审稿人。先后主持国家、省部级项目以及企业重大横向项目20多项,发表国际学术论文100多篇,已授权国家发明专利20多项。

















No comments yet.